
Болезнь Шарко-Мари-Тута
Болезнь Шарко-Мари-Тута относится к группе разнородных генетических заболеваний, для которых характерно поражение периферической нервной системы с развитием атрофии мышц конечностей. Наряду с этим происходит потеря чувствительности, снижение сухожильных рефлексов и деформация конечностей. Заболевание генетически разнородно, к его возникновению может привести около 40 мутаций, локализованных более чем в 20 генах, однако чаще всего встречаются мутации в генах PMP22, MPZ, GJB1, MFN2. Наследование чаще происходит по аутосомно-доминантному типу, но возможны и другие варианты, в том числе и сцепление с Х-хромосомой.
Патогенетические аспекты болезни Шарко-Мари-Тута и её симптомы
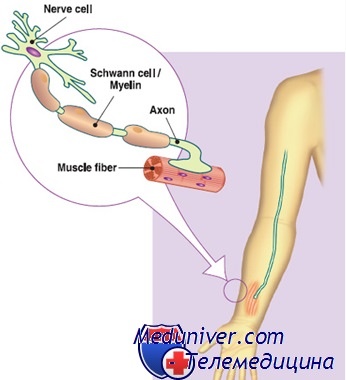
В основе патогенеза болезни Шарко-Мари-Тута лежит повреждение нервов, которое приводит к вторичной мышечной атрофии. Чаще всего страдают «быстрые» двигательные нервные волокна, иннервирующие удаленные мышцы конечностей, берущие на себя большую физическую нагрузку — мышцы стопы и голени. Чуть меньше и позже повреждаются мышцы кистей и предплечий. Также поражаются и чувствительные нервы, что приводит к утрате тактильной, болевой и температурной чувствительности конечностей.
Обычно болезнь Шарко-Мари-Тута манифестирует в возрасте 10–20 лет. Сначала возникает симметричная слабость в ногах, которая приводит к характерному изменению походки по типу степпажа, или, как её еще называют, «петушиной». Затем стопы начинают подворачиваться и деформироваться, увеличивается их свод и образуется полая стопа. По мере прогрессирования атрофии мышц ноги приобретают вид перевернутых бутылок.
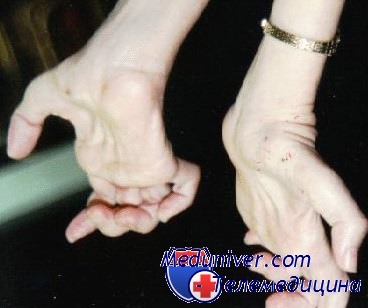
Постепенно присоединяется поражение рук: из-за атрофии мышц кисти она становится похожей на лапу обезьяны. Кроме того, страдает чувствительность, в первую очередь поверхностная. Иногда наблюдается цианоз пораженных конечностей и отеки на них.
Болезнь Шарко-Мари-Тута имеет медленное течение. Иногда период между манифестацией и атрофией рук длится 10 лет. Даже несмотря на развившуюся атрофию, пациенты довольно длительное время сохраняют способность к работе и самообслуживанию. При должном уходе продолжительность жизни не страдает и остается на общепопуляционном уровне.
Классификация болезни Шарко-Мари-Тута
Генетическая классификация очень обширна, поскольку в основе развития заболевания лежат около 40 мутаций, затрагивающих более 20 различных генов. По морфологическим признакам и электромиографическим данным выделяют три основных типа болезни Шарко-Мари-Тута:
- демиелинизирующий тип. Для него характерно разрушение миелиновой оболочки и, как следствие, снижение скорости проведения импульса (СПИ) по срединному нерву;
- аксональный тип. Характерна нормальная или несколько сниженная СПИ по срединному нерву, поскольку в первую очередь страдают аксоны;
- промежуточный тип. Скорость проведения импульса имеет пограничные значения.
Такая классификация имеет смысл для сужения круга поиска при генетической диагностике, поскольку для определенных мутаций характерны свои клинические проявления.
Диагностика болезни Шарко-Мари-Тута
Заподозрить невральную амиотрофию можно на основании вышеперечисленных клинических симптомов. Для дифференциальной диагностики применяется электромиография и электронейрография, а также ряд лабораторных анализов, позволяющих исключить другие нервно-мышечные патологии.
Всем пациентам с характерной клинической картиной показана молекулярно-генетическая диагностика с поиском наиболее характерных для болезни Шарко-Мари-Тута мутаций.
Лечение болезни Шарко-Мари-Тута
Методов излечения болезни Шарко-Мари-Тута на сегодняшний день не разработано. Применяется симптоматическая терапия, направленная на улучшение мышечной трофики. С этой целью назначаются витамины, АТФ, глюкоза, кокарбоксилаза, а также может применяться физиотерапевтическое лечение, ЛФК, массажи. В некоторых случаях необходимо ортопедическое и санаторно-курортное лечение.
В медико-генетическом центре «Геномед» проводится поиск большинства мутаций, приводящих к развитию болезни Шарко-Мари-Тута. В качестве метода исследования мы применяем секвенирование последовательности ДНК.
Болезнь Шарко Мари Тута – проявление патологии
Этиология и встречаемость болезни Шарко-Мари-Тута. Болезнь Шарко-Мари-Тута — генетически разнородная группа наследственных нейропатий, характеризующихся хронической моторной и сенсорной полинейропатией. Данное заболевание подразделяют согласно типам наследования, неврологическим изменениям и клиническим симптомам.
По определению, болезнь Шарко-Мари-Тута 1-го типа — аутосомно-доминантная демиелинизирующая нейропатия; ее распространенность приблизительно 15 на 100 000, она также генетически разнородна. Болезнь Шарко-Мари-Тута 1А типа, представляющая 70-80% всех случаев патологии, вызывается повышенной дозой белка PMP22 из-за дупликации гена PMP22 в хромосоме 17. Новые дупликации составляют 20-33% случаев болезни Шарко-Мари-Тута 1А типа; более 90% этих мутаций возникает в ходе мужского мейоза.
Патогенез болезни Шарко-Мари-Тута
Белок PMP22 — внутримембранный гликопротеид. В периферической нервной системе PMP22 обнаруживают в только в компактном миелине. Функция PMP22 объяснена не полностью, но имеются подтверждения, что он играет ключевую роль в уплотнении миелина.
Доминантные мутации с утратой функции в гене PMP22 и увеличение дозы PMP22 вызывают демиелинизирующую периферическую полинейропатию. Увеличение дозы белка PMP22 возникает при тандемной дупликации участка р11.2 в хромосоме 17. Этот участок размером 1,5 мегабазы ограничен повторяющимися последовательностями ДНК, идентичными почти на 98%. Нарушение спаривания фланговых повторяющихся элементов в ходе мейоза может приводить к неравному кроссинговеру и образованию одной хроматиды с дупликацией региона в 1,5 мегабазы и другой с реципрокной делецией. [Реципрокная делеция вызывает наследственную нейропатию с параличами сдавливания].
Индивидуум, унаследовавший хроматиду с дупликацией, будет иметь три копии нормального гена PMP22 и, таким образом, избыточную экспрессию белка PMP22.
Избыточная экспрессия белка PMP22 или экспрессия его доминантных отрицательных форм приводит к невозможности формирования и поддержки компактного миелина. Образцы биопсии нервов от сильно пораженных детей демонстрируют рассеянную скудность миелина, в биоптатах нервов пациентов при меньшей степени поражения видны участки демиелинизации и гипертрофии миелиновой оболочки. Механизм образования данных изменений при избыточной экспрессии белка PMP22 остается неясным.
Слабость и атрофия мышц при болезни Шарко-Мари-Тута 1-го типа происходят в результате их денервации, вызванной дегенерацией аксонов. Длительные наблюдения за больными показывают возраст-зависимое уменьшение плотности нервных волокон, согласующееся с развитием симптомов болезни. Кроме того, исследования на мышиных моделях указывают, что миелин необходим для функционирования цитоскелета аксонов. Как демиелинизация изменяет цитоскелет аксонов и влияет на их дегенерацию, полностью не объяснено.
Фенотип и развитие болезни Шарко-Мари-Тута
Болезнь Шарко-Мари-Тута 1А типа имеет почти полную пенетрантность, хотя тяжесть, возраст начала и течение болезни заметно изменяются внутри семей и между семьями. Большинство больных не обращаются за медицинской помощью, или не замечая симптомов, или поскольку эти симптомы легко переносятся. С другой стороны, многие имеют тяжелую болезнь, обнаруживаемую в раннем детстве.
Симптомы болезни обычно появляются в первые два десятилетия жизни; начало после 30 лет бывает редко. Обычно симптомы начинаются с незаметной медленно прогрессирующей слабости и атрофии дистальных мышц ног и легкого сенсорного ухудшения. Слабость в стопах приводит к аномалии походки, хлопающей стопе и, в конечном счете, изменению формы стопы (полая стопа молокообразные пальцы стоп) и утрате равновесия; но это редко приводит к утрате способности ходить.
Слабость важных мышц рук обычно появляется в конце болезни и, в серьезных случаях, вызывает аномалию кисти в виде сжатого кулака из-за дисбаланса между мышечной силой сгибателей и разгибателей кисти. Другие связанные симптомы — снижение или отсутствие рефлексов, атаксия и тремор верхних конечностей, сколиоз и пальпаторно утолщенные поверхностные нервы. Иногда также поражаются диафрагмальный и автономный нервы.
При электрофизиологических исследованиях патогномоничный признак болезни Шарко-Мари-Тута 1А типа — равномерное снижение скорости проведения во всех нервах и нервных сегментах в результате демиелинизации. Выраженное снижение скорости проведения обычно присутствует уже к 2-5 годам жизни, хотя клинически явные симптомы могут не обнаруживаться в течение многих лет.
Особенности фенотипических проявлений болезни Шарко-Мари-Тута:
• Возраст начала: от детства до зрелого возраста
• Прогрессирующая дистальная слабость
• Дистальная гипотрофия мышц
• Гипорефлексия
Лечение болезни Шарко-Мари-Тута
Хотя болезнь Шарко-Мари-Тута 1-го типа можно заподозрить на основе клинических, электрофизиологических и патогистологических признаков, окончательный диагноз часто зависит от обнаружения мутации. Воспалительные периферические нейропатии часто трудно отличить от болезни Шарко-Мари-Тута 1-го типа и наследственной нейропатии с параличами сдавливания, и ранее, до появления молекулярной диагностики, многих больных с унаследованной нейропатией лечили иммуносупрессорами, в итоге получая повышенную смертность без улучшения нейропатии.
Лечение симптоматическое, поскольку радикальной терапии к настоящему времени не разработано. Параллельно развитию болезни терапия обычно состоит из трех этапов: укрепляющие и растягивающие упражнения для поддержания ходьбы и других двигательных функций, использование ортопедической обуви и специальных шин и ортопедическая хирургия. При дальнейшем ухудшении могут понадобиться передвижные опоры, такие как костыли, ходунки или, в редких тяжелых случаях, инвалидное кресло. Всем больным необходимо рекомендовать избегать воздействия нейротоксичных медикаментов и химических веществ.
Риски наследования болезни Шарко-Мари-Тута
Поскольку дупликация и большинство точковых мутаций в гене PMP22 аутосомно-доминантные и полностью пенетрантные, каждый ребенок больного родителя имеет 50% шанс развития болезни Шарко-Мари-Тута 1А типа. Тем не менее переменная экспрессивность дупликации и мутаций в гене PMP22 делает невозможным прогноз тяжести болезни.
Пример болезни Шарко-Мари-Тута. В течение нескольких последних лет 18-летняя женщина обращала внимание на прогрессирующую слабость, снижение выносливости и способности бегать и ходить. Она также жаловалась на частые судороги ног, усиливающиеся при охлаждении, и появившиеся затруднения при перешагивании через предметы или подъеме по ступенькам. Она не помнила предшествующих болезней или жалоб, напоминающих воспалительный процесс, например мышечных болей, лихорадки, или ночных потов.
Ни у кого из других членов семьи не было аналогичных проблем или нервно-мышечных заболеваний. При обследовании у пациентки обнаружены худые атрофичные ноги, больше в дистальных отделах, небольшая слабость при сгибании и разгибании стопы, отсутствие рефлексов стопы, снижение коленных рефлексов, «хлопающая» походка и утолщение перонеальных нервов. Ей трудно ходить на пальцах ног и невозможно ходить на каблуках. В остальном результаты обследования оказались нормальными. Как часть обследования, невролог запросил несколько анализов, включая скорость проведения по нервам.
Скорость проведения по нервам оказалась аномальной; средний показатель составил 25 м/с (в норме >43 м/с). Результаты последующей биопсии нерва показали сегментарную демиелинизацию, гипертрофию миелиновой оболочки (избыток шванновских клеток вокруг нервных волокон) и никаких признаков воспаления. Невролог заключил, что эти результаты сильно напоминают демиелинизирующую нейропа-тию, например болезнь Шарко-Мари-Тута 1-го типа, также известную как наследственная мотосенсорная нейропатия 1-го типа. Объяснив, что наиболее частая причина болезни Шарко-Мари-Тута — дупликация в гене периферического миелинового белка 22 (PMP22), невролог предложил анализ на эту дупликацию. Тест подтвердил, что пациентка имела удвоение аллеля PMP22 и болезнь Шарко-Мари-Тута 1А типа.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Болезнь Шарко Мари Тута
Говоря о полой стопе нельзя пройти мимо основной наследственной причины этой патологии – болезни Шарко Мари Тута. Другое её название – наследственная моторная сенсорная нейропатия, различают две формы первая с превалированием мышечной слабости, вторая приводящая к нарушению чувствительности. Является наиболее частым наследственным неврологическим заболеванием (частота 1:2500). При этом заболевании нарушается иннервация таких периферических мышц как: короткая малоберцовая мышца, передняя большеберцовая мышца, собственные мышцы стопы и кисти.

Своим витиеватым названием наследственная моторная сенсорная нейропатия обязана трём врачам: Jean-Martin Charcot (1825–1893), Pierre Marie (1853–1940) Howard Henry Tooth (1856–1925). Их фамилии и легли в основу названия болезни Charcot – Marie – Tooth.
Болезнь Шарко Мари Тута чаще наследуется по аутосомно-доминантному принципу. Это значит, что 50 % детей наследуют заболевание от больного родителя. Также встречается аутосомно-рецессивный тип наследования и сцепленный с X-хромосомой.
Болезнь Шарко Мари Тута связана с мутациями приводящими к синтезу дефективных нейронных белков, большинство мутаций затрагивают структуру миелина, важнейшего белка выступающего в качестве «изоляционной обмотки» нервов, из-за нарушения его структуры скорость передачи сигнала значительно падает. В 60-70 % случаев заболевания мутация происходит в 17 хромосоме, приводя к удвоению участка с геном Р22. Существуют и более редкие формы заболевания, когда мутация происходит в митохондриальном гене MFN2, что приводит к нарушению аксонального транспорта и синаптической передачи.
Таким образом можно выделить две основные формы болезни, в первом случае происходит просто нарушение функции нервов, во втором их дегенерация.
Ортопедические проявления болезни Шарко Мари Тута – полая стопа, молоткообразные пальцы, дисплазия тазобедренных суставов, сколиоз.
Классификация болезни Шарко Мари Тута.
I А тип болезни ШМТ – демиелинизирующее заболевание, сопровождающееся значительным снижением скорости проведения нервных импульсов. Аутосомно-доминантное заболевание, проявления появляются в первой-второй декаде жизни, чаще всего приводит к формированию полой стопы.
II тип ШМТ – постепенное отмирание аксонов за счёт их дегенерации. Имеет более мягкое течение, начинается во второй-третей декаде жизни, чаще приводит к плоской стопе.
Симптомы болезни Шарко Мари Тута.
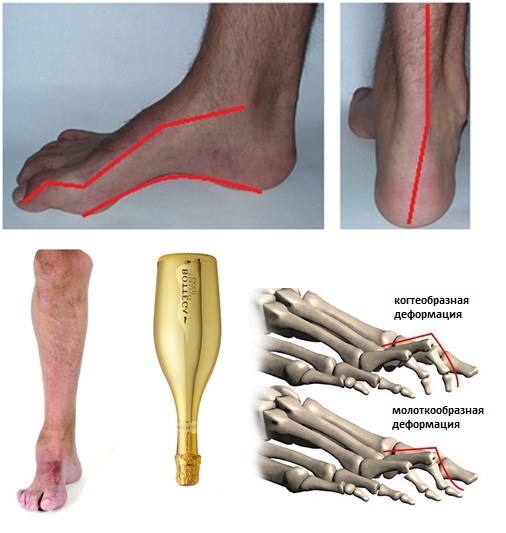
Симптомы болезни Шарко Мари Тута в зависимости от формы заболевания начинаются на втором-третьем десятилетии жизни. В начале заболевания чаще проявляет себя как слабость мышц голени и отвисание стопы. В дальнейшем формируется характерная картина полой стопы, молоткообразная деформация пальцев, инверсия пяточной кости, мышечная атрофия в нижней части голени часто приводит к тому что голень выглядит как «перевернутая бутылка шампанского». Многие пациенты отмечают также слабость кистей и предплечий.
Снижение чувствительности к касанию в стопах, голеностопе и голени постепенно прогрессирует, сопровождается болезненными мышечными спазмами, интенсивность которых значительно варьирует в зависимости от активности процесса. Физическая нагрузка часто провоцирует онемение, спазмы и боль в стопах и кистях. В более тяжёлых случаях болезнь может затрагивать челюстные мышцы, голосовые связки, околопозвоночные мышцы, сопровождаясь нарушениями произношения и сколиозом. Стресс, беременность, длительная иммобилизация приводят к обострению заболевания.
Суммируя наиболее часто встречаемые симптомы: боль по наружному краю стопы, снижение тактильной чувствительности, хромота, частые травмы голеностопного сустава, проблемы с ходьбой по лестнице.
Диагностика болезни Шарко Мари Тута.
При физическом осмотре в первую очередь обращают внимание на деформацию стопы.
Первоначально определяется смещение первого луча в подошвенную сторону. Вторым элементом появляется кавусная деформация за счёт того, что длинная малоберцовая мышца (нормальная) оказывается сильнее поражённой передней большеберцовой мышцы. Третьим элементом деформации становится варусная деформация, которая появляется за счёт того, что задняя большеберцовая мышца оказывается сильнее поражённой короткой малоберцовой мышцы.
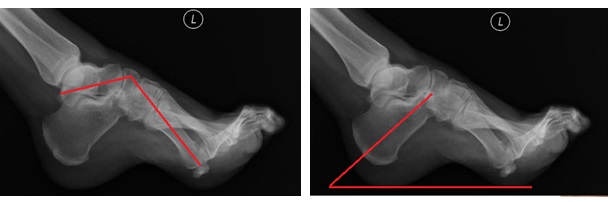
Вторым важным моментом является определение атрофии короткого разгибателя пальцев и короткого разгибателя большого пальца стопы, атрофии голени, особенно в нижнем её отделе, слабость тыльной флексии и эверсии стопы, снижение рефлексов нижней конечности.
Также выполняется тест Колмана (Coleman block test) для определения эластичности заднего отдела стопы.
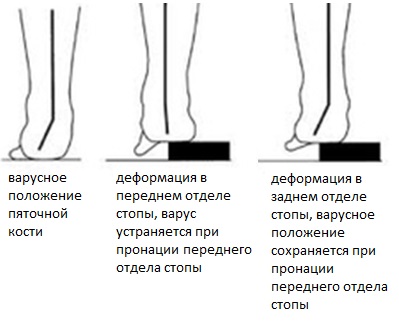
При подкладывании дощечки под наружный край стопы при элдастичной деформации заднего отдела происходит её коррекция до нейтрального положения, в случае ригидной деформации положение пяточной кости остаётся неизменным.
При осмотре верхних конечностей отмечается атрофия собственных мышц кисти.
ЭМГ – снижение скорости проведения нервного возбуждения в малоберцовом, локтевом, срединном нервах.
Генетическое обследование – ПЦР для определения мутации в гене PMP 22, хромосомный анализ для выявлении дупликации плеча 17 хромосомы при самой распространённой аутосомно-доминантной форме заболевания.
С принципами консервативного и хирургического лечения деформации стопы при болезни Шарко Мари Тута вы можете ознакомится в статье посвящённой полой стопе.

Никифоров Дмитрий Александрович
Хирургия стопы и голеностопного сустава, коррекция деформаций конечностей, эндопротезирование суставов, артроскопическая хирургия, спортивная травма.
Невральная амиотрофия Шарко-Мари-Тута
Невральная амиотрофия Шарко-Мари-Тута — это прогрессирующее хроническое наследственное заболевание с поражением периферической нервной системы, приводящем к мышечным атрофиям дистальных отделов ног, а затем и рук. Наряду с атрофиями наблюдается гипестезия и угасание сухожильных рефлексов, фасцикулярные подергивания мышц. К диагностическим мероприятиям относятся электромиография, электронейрография, генетическое консультирование и ДНК-диагностика, биопсия нервов и мышц. Лечение симптоматическое — курсы витаминотерапии, антихолинэстеразной, метаболической, антиоксидантной и микроциркуляторной терапии, ЛФК, массажа, физиопроцедур и водолечение.
МКБ-10

Общие сведения
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) относится к группе прогрессирующих хронических наследственных полиневропатий, в которую входят синдром Русси-Леви, гипертрофическая невропатия Дежерина-Сотта, болезнь Рефсума и другие более редкие заболевания.
По различным данным, невральная амиотрофия Шарко-Мари-Тута встречается с частотой от 2 до 36 случаев на 100 тыс. населения. Зачастую болезнь носит семейный характер, причем у членов одной семьи клинические проявления могут иметь различную выраженность. Наряду с этим наблюдаются и спорадические варианты ШМТ. Лица мужского пола болеют чаще, чем женщины.
Причины
На сегодняшний день практическая неврология как наука не располагает достоверными сведениями о этиологии и патогенезе невральной амиотрофии. Проведенные исследования показали, что у 70-80% пациентов с ШМТ, прошедших генетическое обследование, отмечалось дублирование определенного участка 17-й хромосомы. Определено, что невральная амиотрофия Шарко-Мари-Тута имеет несколько форм, вероятно обусловленных мутациями различных генов. Например, исследователи выяснили, что при форме ШМТ, вызванной мутацией кодирующего митохондриальный белок гена MFN2, происходит образование сгустка митохондрий, нарушающего их продвижение по аксону.
Болезнь Шарко-Мари-Тута характеризуется аутосомно-доминантным наследованием с пенетрантностью на уровне 83%. Встречаются также случаи аутосомно-рецессивного наследования.
Патогенез
Установлено, что большинство форм ШМТ связаны с поражением миелиновой оболочки волокон периферических нервов, реже встречаются формы с патологией аксонов — осевых цилиндров проходящих в центре нервного волокна. Дегенеративные изменения затрагивают также передние и задние корешки спинного мозга, нейроны передних рогов, пути Голля (спинномозговые проводящие пути глубокой чувствительности) и столбы Кларка, относящиеся к заднему спинномозжечковому пути.
Вторично, в результате нарушения функции периферических нервов, развиваются мышечные атрофии, затрагивающие отдельные группы миофибрилл. Дальнейшее прогрессирование болезни характеризуется смещением ядер сарколеммы, гиалинизацией пораженных миофибрилл и интерстициальным разрастанием соединительной ткани. В последующем нарастающая гиалиновая дегенерация миофибрилл приводит к их распаду.
Классификация
В современной неврологической практике невральная амиотрофия Шарко-Мари-Тута подразделяется на 2 типа. Клинически они являются практически однородными, однако имеют ряд особенностей, позволяющих провести такое разграничение.
- Невральная амиотрофия I типа характеризуется существенным снижением скорости проведения нервного импульса. Биопсия нерва обнаруживает сегментарную демиелинизацию нервных волокон, гипертрофический рост непораженных шванновских клеток;
- При амиотрофии ШМТ II типа скорость проведения страдает незначительно, анализ биоптата показывает дегенерацию аксонов.
Отмечена связь болезни Шарко-Мари-Тута и атаксии Фридрейха. В отдельных случаях у пациентов с ШМТ со временем отмечаются типичные признаки болезни Фридрейха и наоборот — иногда по прошествии многих лет клиника атаксии Фридрейха сменяется симптоматикой невральной амиотрофии. Некоторыми авторами даны описания промежуточных форм этих заболеваний. Наблюдались случаи, когда у одних членов семьи диагностировалась атаксия Фридрейха, а у других — амиотрофия ШМТ.
Симптомы
Невральная амиотрофия Шарко-Мари-Тута начинается с развития симметричных мышечных атрофий в дистальных отделах ног. Начальные симптомы манифестируют, как правило, в первой половине второго десятилетия жизни, реже в период от 16 до 30 лет. Они заключаются в повышенной утомляемости стоп при необходимости длительно стоять на одном месте. При этом наблюдается симптом «топтания» – чтобы снять утомляемость стоп пациент прибегает к ходьбе на месте.
В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов. Основной симптом, на который пациенты чаще всего сами обращают внимание – приступообразные болезненные сокращения в икроножных мышцах (крампи), усиливающиеся в ночное время или после длительной физической нагрузки.
Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы. Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа. Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок.
Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса.
Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей. Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей.
Для болезни Шарко-Мари-Тута типично медленное прогрессирование симптомов. Период между клинической манифестацией заболевания с поражения ног и до появления атрофий на руках может составлять до 10 лет. Несмотря на выраженные атрофии, пациенты длительное время сохраняют работоспособное состояние. Ускорить прогрессирование симптомов могут различные экзогенные факторы: перенесенная инфекция (корь, инфекционный мононуклеоз, краснуха, ангина, ОРВИ), переохлаждение, ЧМТ, позвоночно-спинномозговая травма, гиповитаминоз.
Осложнения
Невральная амиотрофия Шарко-Мари-Тута характеризуется ранней инвалидизацией. Вследствие прогрессирующей атрофии дистальных отделов конечностей и выраженных нарушений чувствительности больные постепенно теряют способность к самостоятельной ходьбе. Из-за грубых деформаций кистей рук пациенты не могут сами себя обслуживать. Контрактуры суставов нередко требуют хирургической коррекции.
На ранней стадии заболевания слабость в мышцах ног, гипестезия и гипорефлексия приводят к частым падениям, что повышает вероятность травм и переломов. Наиболее грозные неблагоприятные последствия происходят при сочетании болезни Шарко-Мари-Тута и атаксии Фридрейха. К ним можно отнести слепоту, кардиомиопатию, дыхательную недостаточность.
Диагностика
Курацией пациентов занимаются врачи-неврологи и ортопеды. При опросе больного уточняется возраст, в котором начали появляться симптомы (для болезни ШМТ типична манифестация в 15-25 лет). Важное значение имеет семейный анамнез (наличие близкого родственника с этой патологией). Во время общего осмотра обращается внимание на изменение походки, деформацию стоп и кистей.
При неврологическом осмотре отмечается уменьшение тонуса дистальных отделов верхних и нижних конечностей, ослабление или полное отсутствие сухожильных рефлексов (ахилловых, коленных), снижение кожной чувствительности. Для уточнения диагноза проводятся следующие методы исследования:
- ЭНМГ. При электронейромиографии отмечаются признаки аксональной и демиелинизирующей нейропатии – замедление скорости проведения импульса по двигательным нервам, падение амплитуды М-ответов.
- Компьютерная паллестезиометрия. Данная диагностическая процедура позволяет объективно оценить снижение вибрационной чувствительности – наиболее ранний признаки болезни ШМТ.
- Гистология. При гистологическом исследовании биоптата большеберцового нерва обнаруживаются уменьшение количества миелиновых волокон, разрастание соединительнотканных волокон, атрофию миелина.
- ДНК-анализ. Подтверждающий метод исследования, верифицирующий диагноз. Выявляются дупликации гена белка периферического миелина (PMP22) на 17-й хромосоме.
Дифференциальный диагноз невральной амиотрофии Шарко-Мари-Тута необходимо проводить с наследственными нейромышечными заболеваниями (спинальная мышечная атрофия Верднига-Гоффмана, адренолейкодистрофия, болезнь Пелицеуса-Мерцбахера) и приобретенными хроническими полинейропатиями (синдром Гийена-Барре).
Лечение невральной амиотрофии Шарко-Мари-Тута
Медикаментозная терапия
Для прохождения лечения все больные подлежат обязательной госпитализации в стационар. В настоящее время не существует специфической терапии, способной замедлить прогрессирование аксональной дегенерации и демиелинизации. Однако своевременно начатая грамотная и индивидуально подобранная терапия способна значительно улучшить качество жизни пациентов. Из лекарственных препаратов для симптоматического лечения невральной амиотрофии ШМТ применяются:
- Витамины. Для улучшения микроциркуляции и восстановления нервных волокон назначаются инъекции витаминов группы В (В1, В3, В12). К витамину В6 стоит относиться с осторожностью, так как превышение его дозы оказывает нейротоксический эффект. По данным некоторых исследователей, аскорбиновая кислота способна подавлять образование периферического белка миелина (PMP22).
- Миорелаксанты. С целью устранения болезненных мышечных сокращений пациентам рекомендуется прием медикаментов, расслабляющих скелетную мускулатуру – баклофен, толперизон.
- Кальций и витамин Д. Так как примерно 40% больных имеют остеопороз, для уменьшения риска переломов им показаны препараты кальция и витамина Д (холекальциферол).
- Антихолинэстеразные средства. При болезни ШМТ 2 типа для улучшения нервно-мышечной проводимости целесообразно назначение прозерина, галантамина.
Немедикаментозная терапия
Основное внимание уделяется немедикаментозному лечению невральной амиотрофии Шарко-Мари-Тута. Для достижения максимального терапевтического эффекта применяется комплекс следующих мероприятий:
- Электростимуляция. Для усиления нейротрофики, активации метаболизма в паретичных мышцах и проводимости периферических нервов используется направленная подача электрических импульсов.
- ЛФК. С целью повышения мышечного тонуса рекомендуются регулярные занятия лечебной физкультурой. Наиболее эффективно совмещение активных (выполняются самим пациентом) и пассивных (выполняются специалистом) упражнений.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах (в первую очередь нижних конечностей) выполняются различные виды массажа – ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Бальнеотерапия. Грязевые ванны и грязевые аппликации способствуют коррекции нарушений вегетативной нервной системы и замедлению формирования контрактур.
- Ортопедическое лечение. Чтобы предупредить развитие грубых деформаций больным назначается ношение ортопедической обуви. При нестабильности суставов из-за мышечной слабости, для фиксации стоп в заданном положении используются специальные приспособления (ортезы, подтяжки).
Комплексное проведение данных мероприятий позволяет увеличить мышечную силу, исправить нарушения равновесия и походки. Благодаря этому удается повысить бытовую, социальную адаптацию, работоспособность пациентов.
Хирургическое лечение
При выраженных атрофических явлениях и деформации стопы, значительно затрудняющих самостоятельную ходьбу, когда консервативные методы оказываются безуспешными, показаны ортопедические оперативные вмешательства – метатарзальная остеотомия, остеотомия пяточной кости. В некоторых случаях для восстановления опорной функции стопы может понадобиться проведение артродеза.
Экспериментальное лечение
Продолжаются поиски эффективного лекарства для борьбы с невральной амиотрофией Шарко-Мари-Тута. В клинических испытаниях, где пациенты принимали препарат PXT3003 (комбинация малых доз баклофена, налтрексона и сорбитола), были отмечены положительные результаты в виде увеличения мышечной силы, возобновления чувствительности и сухожильных рефлексов.
Рассматривается возможность использования в качестве лечения ингибиторов HDAC6 – ферментов, стимулирующих регенерацию белков цитоскелета нервных клеток. Эксперименты на лабораторных животных показали, что данные вещества способны значительно замедлить прогрессирование демиелинизации и аксональной дегенерации.
Прогноз и профилактика
Невральная амиотрофия Шарко-Мари-Тута – тяжелое инвалидизирующее заболевание. Большинство пациентов утрачивают способность ходить через 15-20 лет после начала появления симптомов. Однако в виду того, что преимущественно поражаются дистальные отделы конечностей, продолжительность жизни больных практически не отличается от таковой в общей популяции.
Летальные исходы в молодом и среднем возрасте наблюдаются при сочетании с атаксией Фридрейха, когда в патологический процесс вовлекается дыхательная мускулатура и миокард. Специфических методов первичной профилактики не существует. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
1. Оценка качества жизни больных с наследственной невропатией Шарко—Мари—Тута в Красноярском крае/ Шнайдер Н.А., Глущенко Е.В., Козулина Е.А.// Бюллетень Сибирской медицины. – 2011.
2. Клинико-генетическая характеристика наследственной невропатии Шарко—Мари—Тута (на примере Красноярского края): Автореферат диссертации/ Глущенко Е.В. – 2011.
3. Ведение и реабилитация пациентов с наследственной невропатией Шарко—Мари—Тута/ Шнайдер Н.А., Глущенко Е.В.// Комплексная реабилитация: наука и практика. – 2010.
4. Наследственная невропатия Шарко—Мари—Тута/ Шнайдер Н.А., Глущенко Е.В., Кантимирова Е.А. и др. – 2010.
Болезнь Шарко
Болезнь Шарко (в литературе еще встречается как «Болезнь Шарко-Мари», «Болезнь Шарко-Мари-Тута», код по МКБ- G60) –генетически обусловленное хроническое заболевание нервной системы, которое характеризуется развитием постоянно прогрессирующей периферической полинейропатией.
Частота диагностирования болезни Шарко-Мари-Тута согласно статистическим данным где-то один случай на две с половиною тысячи пациентов. Первые симптомы появляются в молодом возрасте. Выраженность симптомов и скорость прогрессирования болезни Шарко-Мари разная у каждого пациента. Процент инвалидизации при заболевании очень высок.

Причины заболевания болезнью Шарко:
- Мутация гена PMP22;
- Мутация MPZ;
- Мутация GJB1;
- Мутация MFN и др.
- аутосомно-доминантный (чаще всего);
- аутосомно-рецессивный;
- Х-сцепленный.
Заболевание имеет много форм, вызванные разным видом мутаций. Качество жизни и возможности работоспособности при болезни Шарко-Мари-Тута значительно ухудшаются, но на продолжительности жизни обычно это не сказывается.
Симптомы болезни Шарко связанные с поражением моторных и сенсорных нервных волокон. Диагностика болезни Шарко-Мари заключается в исключении диагнозов, которые могут давать подобную клиническую картину, и в проведении ДНК-диагностики, но учитывая, что не все виды мутаций известны, она не всегда информативна.
Лечение болезни Шарко-Мари-Тута заключается в симптоматической терапии. Специфическое лечение на данный момент все еще находится в стадии разработки.
Юсуповская больница – одно из лучших медицинских учреждений, где лечатся пациенты с болезнью Шарко-Мари-Тута. Несмотря на то, что терапия направлена только на купирование симптомов, неврология стремительно развивается и питается найти способы лечения многих заболеваний, в том числе и болезни Шарко. Ведение пациентов изданной патологией достаточно сложное, ведь клиническая картина разнообразна, симптомы выражены в неодинаковой степени и п.т. Опыт работы специалистов Юсуповской больницы позволяет оказывать качественную и эффективную медицинскую помощь. Доктора следят за клиническими исследованиями, новыми разработками, препаратами, изучают их эффективность. В случае необходимости, диагностика проводится быстро и с использованием нового оборудования. Персонал работает на благо пациента.
Симптомы болезни Шарко
Симптомы болезни Шарка-Мари появляются в молодом возрасте, чаще всего до двадцати лет. Прогрессирует заболевание постепенно, пациенты долгое время сохраняют работоспособность и возможность самообслуживания. Причинами, которые ускоряют развитие заболевания, могут стать вирусные и бактериальные инфекции, воздействия неблагоприятных факторов среды, травматизм, недостаток витаминов и т.п.
По МКБ-10 болезнь Шарко-Мари-Тута кодируется как «G60 Наследственная моторная и сенсорная невропатия», что связано в какой-то мере с клинической картиной.
В начале заболевания беспокоит чрезмерная усталость, невозможность долго стоять на одном месте, чувство онемения, «беганье мурашек», а дальше присоединяется атрофическое поражение мышц стоп, которое чаще носит симметричный характер. При обследовании выявляют выпадение сухожильных рефлексов. Стопа деформируется на столько, что больные не могут ходить, опираясь на пятки. Прогрессируя, атрофический процесс поражает голени и бедра.
Дальнейшее развитие заболевания болезни Шарко-Мари-Тута приводит к втягиванию в процесс и кистей, далее – предплечий. Мышцы шеи, туловища и плечевого пояса не атрофируются. Мышцы проксимальных отделов компенсаторно увеличиваются.
Все виды чувствительности нарушаются, но больше всего страдает поверхностная, особенно температурная.
Диагностика болезни Шарко
- Осмотр неврологом;
- Лабораторные исследования;
- Инструментальные исследования;
- ДНК-исследования.
Полное обследование необходимо для исключения заболеваний, в которых клиническая картина сходна. К ним относят: боковой амиосклероз, миотония, метаболическая невропатия. Для исключения хронических полинейропатий проводят биопсию мышц.
Лечение болезни Шарко
Все способы лечения болезни Шарко-Мари-Тута не радикальны. Симптоматическое лечение включает медикаментозную терапию, физиотерапию, лечение у ортопеда и т.п.
Физиотерапевтическое лечение болезни Шарко-Мари-Тута включает ЛФК, массаж, электрофорез, диадинамотерапию, терапию лечебными грязями, разные виды ванн и др.
Медикаментозная терапия направлена на улучшение питания мышечных волокон. С этой целью назначают кокарбоксилазу, глюкозу, аденозинтрифосфат и др. Так же широко применяют антиоксидантные средства, препараты, улучшающие микроциркуляцию, и витамины. Хорошо зарекомендовали себя препараты, которые тормозят активность ацетилхолинэстеразы и повышают уровень ацетилхолина, например, прозерин, галантамин.
Дальнейшие разработки новых препаратов, направленные на радикальные меры – это мир без болезни Шарко-Мари-Тута. Прогрессирование болезни Шарко-Мари-Тута не отражается на том, сколько живут пациенты.
В Юсуповской больнице специалисты долгие годы помогают пациентам держать болезнь под контролем. Минимальная выраженность симптомов и медленное прогрессирование – результат работы врачей. В комфортных палатах, на новых тренажерах, в хорошо оснащенных кабинетах – вот где проходит лечение болезни Шарко-Мари-Тута. Не затягивайте с лечением, запишитесь на консультацию.
X Международная студенческая научная конференция Студенческий научный форум – 2018

Наследственная невропатия Шарко-Мари-Тута является одной из наиболее частых форм наследственных невропатий (около 80 % случаев). Распространенность невропатии в мире варьирует от 9,17 до 30 человек на 100 000 населения. При этой патологии чаще страдают люди молодого возраста: заболевание обычно развивается на втором десятилетии жизни (12-15 лет), но существуют формы с ранним дебютом (6-7 лет). Чем раньше манифестирует заболевание, тем тяжелее оно протекает. Средняя продолжительность жизни и интеллект пациентов не страдают, но клинические проявления синдрома, наступающие в течение первого десятилетия от дебюта, ограничивают трудоспособность. Осложнения болезни (периферические парезы, контрактуры, расстройства равновесия и др.) приводят к нарушению двигательных и мануальных функций, негативно отражаются на качестве жизни пациентов и ограничивают их ежедневную физическую активность
Цель
Изучение развитие болезни, этиологии, частоты встречаемости и патогенеза, а также лечения.
Задачи
Проанализировать этиологию и встречаемость болезни
Рассмотреть фенотип и развитие болезни
Определить риск наследования
Рекомбинация между повторяющимися последовательностями ДНК
Возраст начало: от детства до зрелого возраста
Прогрессирующая дистальная слабость
Дистальная гипотрофия мышц
Фундаментальные сведения Этиология и встречаемость болезни
Болезнь Шарко – Мари – Тута это генетически разнородная группа наследственных нейропатий, характеризующихся хронической моторной и сенсорной полийропатией. Данное заболевание подразделяют согласно типам наследования, неврологическим изменениям и клиническим симптомам. По определению, болезнь Шарко – Мари – Тута 1 типа – аутосомно – доминантная демиелинизирующая нейропатия; ее распространенность приблизительно 15:100000, она также генетически разнородна. Болезнь Шарко – Мари – Тута 1А типа, представляющая 70-80% всех случаев патологии, вызывается повышенной дозой белка РМР22 из-за дупликации гена РМР22 в хромосоме 17. Новые дупликации составляют 20-33% случаев болезни Шарко – Мари – Тута 1А типа; более 90% этих мутаций возникают в ходе мужского мейоза.
Белок РМР22 – внутримембранный гликопротеид. В ПНС РМР22 обнаруживают только в компактном миелине. Функции РМР22 объяснена не полностью, но имеются подтверждения, что он играет ключевую роль в уплотнении миелина.
Доминантные мутации с утратой функции в гене РМР22 и увеличение дозы РМР22 вызывают демиелинизирующую перефирическую полинейропатию. Увеличение дозы белка РМР22 возникает при тандемной дупликации участка р11.2 в хромосоме 17. Этот участок размером 1,5 мегабазы ограничен повторяющимися последовательностями ДНК, идентичными почти на 98%. Нарушение спаривания фланговых повторяющихся элементов в ходе мейоза может приводить к неравному кроссинговеру и образованию одной хроматиды с дупликацией регионов 1,5 мегабазы и другой реципрокной делецией ( реципрокная делеция вызывает наследственную нейропатию с параличами сдавливания). Индивидуум, унаследовавший хроматиду с дупликацией, будет иметь три копии нормального гена РМР22 и , таким образом, избыточную экспрессию белка РМР22.
Избыточная экспрессия белка РМР22 или экспрессия его доминантных отрицательных форм приводит к невозможности формирования и поддержки компактного миелина. Образцы биопсии нервов от сильно пораженных детей демонстрируют рассеянную скудность миелина, в биоптатах нервов пациента при меньшей степени поражения видны участки демиелинизации и гипертрофии миелиновой оболочки. Механизм образования данных изменений при избыточной экспрессии белка РМР22 остается не ясным.
Слабость и атрофия мышц при болезни Шарко – Мари – Тута 1 типа происходит в результате их денервации, вызванной дегенерацией аксонов. Длительные наблюдения за больными показывают возраст – зависимое уменьшение плотности нервных волокон, согласующееся с развитием симптомов болезни. Кроме того , исследования на мышиных моделях указывают, что миелине необходим для функционирования цитоскелета аксонов. Как демиелинизация изменяет цитоскелет аксонов и влияет на их дегенерацию, полностью не объяснено.
Фенотипы развития болезни
Болезнь Шарко – Мари – Тута 1А типа имеет почти полную пенетрантность, хотя тяжесть, возраст начала и течение болезни заметно изменяются внутри семей и между семьями. Большинство больных не обращаются за медицинской помощью, или не замечая симптомов или поскольку эти симптомы легко переносятся. С другой стороны многие имеют тяжелую болезнь, обнаруживаемую в раннем детстве.
Симптомы болезни обычно появляются в первые два десятилетия жизни; начало после 30 лет бывает редко. Обычно симптомы начинаются с незаметной медленно прогрессирующей слабости и атрофии дистальных мышц ног и легкого сенсорного ухудшения. Слабость в стопах приводит к аномалии походки, хлопающей стопе, и , в конечном счете, изменению формы стопы(полая стопа, молокообразные пальцы стоп) и утрате равновесия; но это редко приводит к утрате способности ходить. Слабость важных мышц рук обычно появляется в конце болезни и , в серьезных случаях, вызывает аномалию кисти в виде сжатого кулака из-за дисбаланса между мышечной силой сгибателей и разгибателей кисти. Другие связанные симптомы – снижение или отсутствие рефлексов, атаксия и тремор верхних конечностей, сколиоз и пальпаторно утолщенные поверхностные нервы. Иногда также поражаются диафрагмальный и автономный нервы.
При электрофизиологических исследованиях патогномоничный признак болезни Шарко – Мари – Тута 1А типа – равномерное снижение скорости проведения во всех нервах и нервных сегментах в результате демиелинизации. Выраженное снижение скорости проведение обычно присутствует уже к 2-5 годам жизни, хотя клинически явные симптомы могут не обнаруживаться в течение многих лет. 1
Хотя Шарко – Мари – Тута 1 типа можно заподозрить на основе клинических, электрофизиологических и патогистологических признаков, окончательный диагноз часто зависит от обнаружения мутации. Воспалительные периферические нейропатии часто трудно отличить от болезни Шарко – Мари – Тута 1 типа и наследственной нейропатии с параличами сдавливания, и ранее, до появления молекулярной диагностики, многих больных с унаследованной нейропатией лечили иммуно-супрессорами, в итоге получая повышенную смертность без улучшения нейропатии.
Лечение симптоматическое, поскольку радикальной терапии к настоящему времени не разработано. Параллельно развитию болезни терапия обычно состоит из трех этапов: укрепляющая и растягивающие упражнения для поддержания ходьбы и других двигательных функций, использование ортопедической обуви и специальных шин и ортопедическая хирургия. При дальнейшем ухудшении могут понадобиться передвижные опоры, такие как костыли, ходунки или, в редких тяжелых случаях , инвалидное кресло. Всем больным необходимо рекомендовать избегать воздействия нейротоксичных медикаментов и химических веществ.
Поскольку дупликация и большинство точковых мутаций в гене РМР22 аутосомно – доминантные и полностью пенетрантные, каждый ребенок больного родителя имеет 50% шанс развития болезни Шарко – Мари – Тута 1А типа.
Список литературы
Роберт Л. Ньюссбаум. Медицинская генетика 492-493 с.
1 Клинический случай: в течение нескольких лет 18-летняя девушка обращала внимание на прогрессирующую слабость, снижение выносливости, частые судороги ног. Ни у кого из членов семьи не было таких симптомов. При обследовании у девушки обнаружены слабость при сгибании и разгибании стопы, отсутствие рефлексов стопы, утолщение перонеальных нервов. Невролог запросил анализ на скорость проведения по нервам и скорость оказалась аномально мала. Результат биопсии показал сегментарную димиелинизацию, гипертрофию миелиновой оболочки. Врач предложил сделать анализ на дупликацию гена РМР22, и анализ оказался положительным, что подтвердило диагноз Шарко – Мари – Тута 1А типа.
Образ жизни при болезни Шарко-Мари-Тута
БОЛЕЗНЬЮ ШАРКО-МАРИ-ТУТА СТРАДАЮТ ОКОЛО 3-Х МИЛЛИОНОВ ЧЕЛОВЕК ВО ВСЕМ МИРЕ. В КРАСНОЯРСКЕ И КРАСНОЯРСКОМ ЭТО НАСЛЕДСТВЕННОЕ ЗАБОЛЕВАНИЕ ПЕРИФЕРИЧЕСКИХ НЕРВОВ ТОЖЕ ДОСТАТОЧНО РАСПРОСТРАНЕНО. В НАШЕМ РЕГИОНЕ ЕГО ВСТРЕЧАЕМОСТЬ ДОСТИГАЕТ 10 СЛУЧАЕВ НА 100 ТЫСЯЧ ЧЕЛОВЕК. ОБРАЗ ЖИЗНИ (ВЫБОР ФИЗИЧЕСКИХ НАГРУЗОК, СПОРТА, СТИЛЯ ПИТАНИЯ) И ДИСПАНСЕРНОЕ НАБЛЮДЕНИЕ У НЕЙРОГЕНЕТИКА, ОРТОПЕДА И ПОДИАТРА МОЖЕТ СУЩЕСТВЕННО ЗАМЕДЛИТЬ ТЕМПЫ ПРОГРЕССИРОВАНИЯ ЗАБОЛЕВАНИЯ И УЛУЧШИТЬ КАЧЕСТВО ЖИЗНИ ПАЦИЕНТОВ.
Что такое болезнь Шарко-Мари-Тута?
Болезнь Шарко-Мари-Тута (синонимы: наследственная нейропатия Шарко-Мари-Тута, ШМТ, невральная амиотрофия) – наследственное заболевание периферической нервной системы, характеризующееся поражением чувствительных и двигательных нервов рук и ног.
Длинные волокна периферических нервов повреждаются сильнее, поэтому в первую очередь нарушаются функции наиболее удаленных (дистальных) отделов конечностей, испытывающих большую физическую нагрузку – это мышцы стоп и голеней. Мышцы кистей и предплечий обычно вовлекаются в патологический процесс в меньшей степени и на более поздних сроках развития заболевания.
Первые симптомы обычно появляются в возрасте 10-20 лет. Болезнь Шарко-Мари-Тута прогрессирует постепенно, исподволь:
- постепенно нарастает слабость в пораженных мышцах;
- становится заметным, что мышцы стоп и голеней уменьшились в объеме (атрофия мышц);
- изменяется форма конечностей: форма ног начинает напоминать вид «перевернутой бутылки шампанского» (так называемые «ноги аиста»);
- формируется сгибательная деформации стоп (сначала стопы могут приобретать высокий свод, затем формируется так называемая «полая стопа»);
- затрудняется ходьба: стоять и ходить на носках и/или пятках становится практически невозможно;
- позднее (чаще примерно через 10 лет после появления первых симптомов) в патологический процесс вовлекаются руки (кисти и предплечья): в них происходят те же изменения, что и в ногах, с формированием деформации по типу «когтистых лап»;
- иногда появляется неконтролируемая дрожь в пальцах кистей (постуральный или постурально-кинетический тремор);
- возможно искривление позвоночника (сколиоз или кифосколиоз) за счет вовлечения в атрофический процесс мышц туловища.
Какие нагрузки следует избегать людям с болезнью Шарко-Мари-Тута?
Детям и взрослым, страдающим наследственной нейропатией Шарко-Мари-Тута (синоним: болезнь Шарко-Мари-Тута), необходимо избегать чрезмерных физических и психических перегрузок, так как это может спровоцировать ухудшение состояния (нарастание слабости в мышцах рук и ног и нарушение чувствительности в конечностях).
Какие виды спорта полезны при болезни Шарко-Мари-Тута?
Дозированные физические нагрузки, такие как лечебная гимнастика, пилатес, катание на велосипеде и плавание, рекомендуются для профилактики развития контрактур суставов нарастания выраженности мышечных атрофий.
Пловцу Донне ДеУвик (Donna DeWick) в 2004 году был установлен диагноз наследственной нейропатии Шарко-Мари-Тута. Пережив тяжелое потрясение в связи с этим сообщением врачей, а также осознав, что в период с 2004 по 2007 годы состояние ее здоровья стало резко ухудшаться, она решила взять под контроль свое здоровье и благополучие, сохранить свою мобильность. Со временем она использовала спорт не только как транспортное средство по сбору средств благотворительности, но и как способ поверить в свои силы. Недавно она участвовала в 7000-метровом заплыве через Большой Чесакпикский залив.
«Независимо от результата, я действительно сделала все возможное, чтобы дать себе наилучший шанс для успеха в этот день», – сказала корреспондентам Донна.
В выборе спортивных нагрузок и спортивной диеты при болезни Шарко-Мари-Тута важен междисциплинарный подход и помощь, как лечащего врача нейрогенетика, так и ортопеда – специалиста по ЛФК и спортивной медицине. Так подход с успехом реализован на базе Неврологического центра Университетской клиники, где детям и взрослым подбирается индивидуальная программа спортивных нагрузок в тренажерном зале, бассейне, а также в домашних условиях в зависимости от характера и степени тяжести заболевания, возраста пациента. Консультирование и сопровождение пациентов совместно с нейрогенетиками центра осуществляет специалист по ЛФК и спортивной медицине Сергей Валентинович Невзоров.
Что необходимо предпринять для профилактики деформаций стоп?
В качестве профилактики развития ранней деформации стоп, необходимо носить удобную, не стесняющую стопы, обувь. Соответствующая обувь является очень важным пунктом для людей, страдающих наследственной нейропатией Шарко-Мари-Тута, но часто они испытывают трудности при поиске подходящей им обуви из-за высокого подъема и специфической формы ноги («полая стопа») и пальцев ног («молоточкообразные пальцы»).
Пациентам также рекомендуется регулярно посещать специалиста по проблеме заболеваний стоп – подиатра, (от лат. слова, означающего ступня) прием которого организован на базе кабинета ‘Диабетической стопы’ Университетской клиники (прием ведет хирург-подиатор, к.м.н. Виктория Павловна Овчинникова). Это важно для предупреждения и своевременного лечения трофических дефектов мягких тканей стопы в местах максимальной травматизации за счет развития деформаций стоп.
Аномалии ходьбы могут быть исправлены путем использования разных типов подтяжек, которые называются AFOs (ankle-foot orthoses). Эти крепления помогают контролировать тыльное сгибание ноги и голени, нестабильность голеностопного сустава и, зачастую, обеспечивают лучшее чувство равновесия. Ортезы позволяют пациентам максимально долго оставаться физически активными, самостоятельно передвигаться, предотвращая падения и получение травм.
Ортезы для фиксации стопы в физиологическом положении используются при синдроме «свисающей стопы».
Екатерина Александровна Козулина, кандидат медицинских наук, нейрогенетик, ведущий специалист по наследственной нейромышечной патологии Неврологического центра Университетской клиники поможет подобрать ортезы в зависимости от индивидуальных особенностей детей, подростков и взрослых, страдающих наследственной нейропатией Шарко-Мари-Тута, а также от выраженности амиотрофий и типа локомоторных нарушений. Некоторые люди для стабильности походки нуждаются в костылях или трости, и менее 5% нуждаются в инвалидных креслах.
Какой образ питания рекомендуется детям и взрослым, страдающим болезнью Шарко-Мари-Тута?
Пациенты должны придерживаться хорошо сбалансированной диеты, чтобы избежать ожирения, которое может способствовать возникновению боли в пояснице и оказывает дополнительную нагрузку на ослабленные мышцы. Пища должна быть богата антиоксидантами (витамины Е, С, А, селен).
Источником витамина С служит растительная пища. Особенно богаты витамином С перец и черная смородина, укроп, петрушка, капуста, щавель, цитрусовые, земляника. Но чемпионом среди всех растений является шиповник – 1,2 г на 100 г сухих ягод. Ежесуточно следует получать 50 мг аскорбиновой кислоты, однако наиболее оптимальной (вне стрессовой ситуации) является доза 100-200 мг в сутки. При ШМТ суточная доза витамина С может быть увеличена до 2 г. По данным зарубежных исследований, ежедневное употребление аскорбиновой кислоты в дозе 1-3 г способствует снижению сверхэкспрессии патологического гена РМР22 и уменьшает фенотипические проявления заболевания [Passage E. et al., 2004].
Считается что, аскорбиновая кислота в суточной дозе 100 мг достаточна для насыщения клетки крови, 400 мг/сут – достаточна для насыщения плазмы. M. Fontes предложил использование больших доз аскорбиновой кислоты (до 3 г/сут), объясняя это тем, что только большие концентрации эффективны на уровне клетки при ШМТ.
Может ли прием лекарств негативно влиять на тяжесть заболевания?
Людям, страдающим наследственной нейропатией Шарко-Мари-Тута, необходимо избегать приема лекарственных препаратов, оказывающих токсическое действие на периферическую нервную систему, нарушающих функцию пораженных при ШМТ нервов [Weimer W. et al., 2006]. http://www.krasmedic.ru/article/?idc=50&ids=&ida=615
Авторы: Наталья Алексеевна Шнайдер, заведующая кафедрой медицинской генетики и клинической нейрофизиологии ИПО КрасГМУ им. проф. В.Ф. Войно-Ясенецкого, руководитель Неврологического центра Университетской клиники, д.м.н., проф.;
Екатерина Александровна Козулина, к.м.н., невролог-нейрогенетики Неврологического центра Университетской клиники
Елена Владимировна Глущенко, к.м.н., ассистент кафедры, невролог-нейрогенетик
Источники: – Шнайдер Н.А., Глущенко Е.В., Кантимирова Е.А., Козулина Е.А. Наследственная нейропатия Шарко-Мари-Тута: Учебное пособие для последипломного образования врачей. – Красноярск: Издательство «Гротеск», 2010. – 105 с.
– Всё про гены http://vse-pro-geny.com/ru_home.html
– Блог Донны ДеУвик «Я ПЫТАЮСЬ, потому что я МОГУ ….» (I TRI because I CAN…) http://www.beatinglimitations.com/
-National CMT resource center http://help4cmt.com/factsheets/?t=exercise
©Авторские права защищены. При использовании материала ссылки на авторов, сайт Университетской клиники и КрасГМУ им. проф. В.Ф. Войно-Ясенецкого обязательны.

