
Синдром Фабри — редкое генетически детерминированное заболевание
 В мире насчитываются тысячи болезней, которые являются редкими и относятся к генетическим мутациям. К таким заболеваниям относится болезнь Фабри. Все аспекты об этой болезни рассмотрены в статье ниже.
В мире насчитываются тысячи болезней, которые являются редкими и относятся к генетическим мутациям. К таким заболеваниям относится болезнь Фабри. Все аспекты об этой болезни рассмотрены в статье ниже.
Что это за заболевание
 Заболевание имеет двойное название Андерсона-Фабри. При развитии у больного обнаруживают сгустки гликосфинголипидов, один из видов жиров, в человеческих клетках. Поражается целый ряд органов: сердце, почки, нервные клетки и органы зрения. Отличительной особенностью болезни является появление на коже специфических высыпаний.
Заболевание имеет двойное название Андерсона-Фабри. При развитии у больного обнаруживают сгустки гликосфинголипидов, один из видов жиров, в человеческих клетках. Поражается целый ряд органов: сердце, почки, нервные клетки и органы зрения. Отличительной особенностью болезни является появление на коже специфических высыпаний.
Жизнь с таким заболеванием сложна. Продолжительность жизнедеятельности человека сокращается. Сегодня специалисты борются с пороком при помощи заместительного лечения.
Открыли болезнь 2 доктора с фамилиями Фабри и Андерсон в конце 19 века. На 300 тысяч рождающихся детей 1 младенец появляется с данной патологией.
Болезнь Фабри связывают с дефектами лизосом. А-галактозидаза, фермент лизосом, прекращает вырабатываться, при этом гликосфинголипиды с трудом расщепляются и продукты метаболизма накапливаются на внутренних органах и их тканях. Вся система органов перестает нормально работать. Больше всего страдают сосуды и сердечные мышцы, состоящие из гладкомышечной ткани.
Что провоцирует синдром Фабри
 Синдром Фарби в основном спровоцирован мутацией половой Х-хромосомы. А-галактозидаза имеет свой код, который находится на одном из участков вышеописанной хромосомы. Если возникает дефект на этом месте, то выработка фермента идет на снижение.
Синдром Фарби в основном спровоцирован мутацией половой Х-хромосомы. А-галактозидаза имеет свой код, который находится на одном из участков вышеописанной хромосомы. Если возникает дефект на этом месте, то выработка фермента идет на снижение.
Мутационное нарушение передается через рецессивный признак. Следуя информации из курса школьной биологии, если мужской пол наследует мутагенную Х-хромосому, то он однозначно будет болен синдромом Фарби. Однако дальнейшее потомство предполагает следующее: девочки в 100% случаев родятся больными, а мальчики здоровыми.
Другая картина обстоит с заболеванием, проявляющимся у женщин. Женский пол имеет 2 Х-хромосомы. Если одна из них с дефектом, то болезнь проявит себя спокойно. Если обе строение обоих хромосом нарушено, то болезнь будет активно прогрессировать, но это встречается крайне редко. Сыновья и дочери больной женщины в 50% случаев наследуют патологию.
Симптомы вызванные болезнью
Болезнь Андерсона появляется обычно в детском возрасте от 1 года. Взрослые люди болеют в редких случаях. Список возможных симптомов:
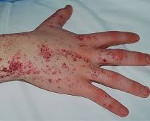
- раздражение на кожном покрове рук, ног, паховой области и лице, которое появляется в виде язвочек, при контакте с горячими предметами область поражений увеличивается;
- прекращение работы потовых желез;
- сильная слабость после легких физических нагрузок;
- лихорадка, повышение температуры;
- появление ревматического синдрома суставов;
- преимущественно у детей появление вегетативных нарушений;
- боли в мышцах по мере прогрессирования заболевания;
- упадок зрения, может развиться катаракта, разрушаются сосуды сетчатки глаза;
- сердечные боли;
- проблемы с почками;
- болезненные ощущения при сгибании и разгибании конечностей, может длиться несколько часов или неделями;
- после плотного обеда и ужина заболевает желудок или кишечник;
- тошнота;
- рвота;
- диарея.
К симптомам также относят внешние проявления болезни. Обычно внешние изменения заметны в возрасте 13 лет. Типичные характеристики у мальчиков:
- западение переносицы;
- выступление бровей и лба вперед;
- в 2 раза увеличенная нижняя челюсть;
- в 2 раза увеличивается рот;
- расширяются губы.
Такие изменения вызывает соматотропный гормон.
Болезнь сопровождается болью, которая подразделяется на 2 типа:
- Боль, возникающая как на физический раздражитель, называемая нейропатической. При любом прикосновении к ступням и ногам, больной чувствует жжение и покалывания.
- Внезапные боли. Появляются внезапно в виде приступов. Зачастую болевой порок возникает на ступнях и ногах, иногда в других частях тела.
Диагностика при данном синдроме
Чтобы диагностировать синдром Фабри, для начала выявляется насколько активен вышеуказанный фермент и наличие жировых отложений на внутренних органов. Анализы женщин и мужчин отличаются друг от друга.
Следующий список описывает ряд исследований для женщин:
- ДНК-анализ.
- Проверка родственных связей.
- Внешний осмотр на наличие красных язвочек.
Беременных женщин также обследуют на возможность заболевания. Это называется пренатальной диагностикой. Специалисты проводят исследование плода, так как после рождения признаков синдрома Фабри до 1 года не наблюдается.
Рекомендации по лечению
Лечат болезнь Фабри-Андрсона несколькими способами. Подробно все способы лечения симптоматических проявлений рассмотрены в таблице ниже:
| Симптом | Как лечат | |
|---|---|---|
| Боли и заболевания в почках | Гипертензия | Контролируется артериальное давление. Назначают ингибиторы АПФ. |
| Желудочные боли | Назначается диета, при которой исключаются из рациона жиры. Больной питается понемногу и 6 раз в день. Врач назначает стимуляторы моторики. | |
| Боли в сердце и сосудах | Врач назначит ингибиторы АПФ, блокаторы кальциевых каналов, диуретики и бетта-блокаторы. |
Профилактика заболевания
Чтобы предостеречь себя и детей от заболевания Фабри, необходимо проводить профилактические меры. Для этого рекомендуется посещать всех врачей, не реже одного раза в год. Для предотвращения болезни у детей следует посещать педиатра при любых подозрениях на болезнь, а также проводит пренатальное обследование плода.
Чтобы болезнь не прогрессировала, необходимо постоянное лечение и наблюдение у лечащего врача. Необходимо также следить за развитием ребенка, если не проводилось пренатальное обследование: первые признаки синдрома Фабри заметны у ребенка в пятилетнем возрасте. Ярким симптомом болезни в детстве считается появление красных язвочек на теле.
Генетические заболевания
Патофизиология
Дефицит фермента альфа-галактозидазы А (а-GAL А, кодируется геном GLA), который возникает из-за мутации, вызывает накопление гликолипида известного как глоботриаосилцерамид (сокращенно GB3, GL-3) в кровеносных сосудах, других тканях и органах. Такое накопление приводит к нарушению их нормального функционирования.
Мутации ДНК, которые являются причиной возникновения заболевания, наследуются по рецессивному Х-сцепленому типу. Болезнь поражает гемизиготних мужчин (т.е. всех мужчин), а также гомозиготных и, во многих случаях гетерозиготных, женщин. В то время как у мужчин обычно проявляются серьезные симптомы, для женщин характерна вариация течения заболевания, оно может быть бессимптомным или с проявленем тяжелых симптомов. Эта изменчивость, как полагают, связана с Х-инактивационой моделью во время эмбрионального развития женщины.
Симптомы
Симптомы заболевания, как правило, впервые проявляются в раннем детстве и обычно их очень трудно понять. В связи с тем, что болезнь Фабри редко встречается в практике врачей это иногда приводит к установлению неверного диагноза. Проявления болезни, как правило, с возрастом человека ухудшаются.
Боль
Боль во всем теле или локализованные боли в конечностях (известный как акропарестезия) или желудочно-кишечном тракте (ЖКТ) является характерным явлением для пациентов с болезнью Фабри.
Распространенным и серьезным следствием болезни являются осложнения, связанные с почками. Почечная недостаточность (уремия) может ухудшаться со временем. Протеинурия (которая может вызвать пенистость мочи) часто является первым признаком поражения почек. Конечная стадия почечной недостаточности у мужчин, как правило, наступает в 30-40 летнем возрасте, и является частой причиной смерти.
Проявления со стороны системы кровообращения
Сердечные осложнения возникают при накоплении гликолипидов в клетках сердца. Влияние на сердце с возрастом ухудшается, что может привести к увеличению риска возникновения сердечно-сосудистых заболеваний. Часто у больных наблюдается гипертония (высокое кровяное давление) и метаболические изменения (кардиомиопатия).
Дерматологические проявления 
Ангиокератомы (маленькие, безболезненные папулы, которые могут появляться на любой части тела, но обычно появляются на бедрах, вокруг пупка, ягодицах, нижней части живота и в паху) тоже является распространенным симптомом.
Ангидроз (отсутствие потоотделения) является распространенным симптомом в отличие от гипергидроза (повышенная потливость), который встречается значительно реже.
Кроме того, у пациентов могут возникать симптомы подобные синдрому Рейно вместе с нейропатией (в частности, жгучая боль в конечностях).
Поражение глаз
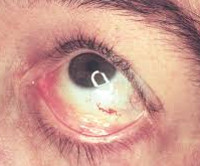
 При заболевании характерны также поражение глаз, а именно помутнение роговицы (известное как кератопатия). Кератопатия может проявляться и у бессимптомных носителей, но необходимо исключить любые другие причины ее появления (например, осадок препарата в роговице). Это помутнение не влияет на остроту зрения.
При заболевании характерны также поражение глаз, а именно помутнение роговицы (известное как кератопатия). Кератопатия может проявляться и у бессимптомных носителей, но необходимо исключить любые другие причины ее появления (например, осадок препарата в роговице). Это помутнение не влияет на остроту зрения.
Другие симптомы включают конъюнктивитный аневризмм, заднюю субкапсунальную катаракту, отек диска зрительного нерва, отек макулы, атрофию зрительного нерва и дилатацию сосудов сетчатки.
Другие проявления
Другими общими симптомами заболевания являются: усталость, невропатия (в частности, жгучая боль в конечностях), цереброваскулярные эффекты, приводящие к повышенному риску возникновения инсульта, шум в ушах (звон в ушах), головокружение, тошнота, неспособность набрать вес, электролитный дисбаланс, диарея и другие проявления.
Диагноз
Болезнь Фабри диагностируется в том случае, если присутствуют присущие болезни симптомы, а также после анализа крови с помощью которого определяют уровень активности альфа-галактозидазы. Однако, при установлении диагноза женщинам-носителям, он может вводить в заблуждение в связи с случайной природой X-инактивации. Хромосомный анализ гена GLA является наиболее точным методом диагностики, в связи с возможностью определения многих мутаций, вызывающих заболевания. Почечная биопсия может также указать на наличие болезни Фабри, если при обследовании будут определены избыточные накопления липидов.
Естественно, что альфа-галактозидаза А (а-GAL А), вероятно, будет присутствовать только на очень низком уровне в крови, особенно у мужчин. У женщин в связи с моделью Х-инактивации уровень а-GAL А, как правило, является нормальным, даже если у пациента проявляются специфические симптомы. Sifap (инсульт у молодых пациентов) – проект, в котором будут исследовать связь между инсультом и болезнью Фабри.
Ошибочно установлен диагноз болезни Фабри
Педиатры равно как и терапевты, часто ошибаются при диагностировании болезни Фабри.
Лечение
До 2000-го года, лечения болезни Фабри было направлено на преодоление симптомов, возникающих при прогрессирования заболевания.
В 2001 году начали применяться три направления ферментнозаместительной терапии (ФЗТ): альфа галактозидазы (Реплагал (Replagal), производство Shire) и бета галактозидазы (Фабразим (Fabrazyme), производство Genzyme).
Лечение путем замены дефицитного фермента осуществляется инъекциями каждые две недели и является наиболее применяющимся методом. Стоимость этих препаратов – высокая (примерно $ 250,000 США в год / пациента) и остается непреодолимым барьером для многих пациентов в некоторых странах. Инфузия может осуществляться самим пациентом, медсестрой дома у больного или в медицинском учреждении. Ферментозаместительная терапия не является панацеей, но может помочь нормализовать обмен веществ и предотвратить прогрессирование заболевания, а также избежать повторения симптомов.
Боль при болезни Фабри тоже утоляется благодаря ФЗТ, однако схемы лечения болевого синдрома могут также включать применение анальгетиков, противосудорожных, и нестероидных противовоспалительных препаратов.
Упоминания о болезни Фабри в поп-культуре:
– Доктор Хаус («Полный провал», 2 серия 6 сезона (эпизод 113)) центров для пациентов с болезнью Фабри;
– «Клиника» (My Catalyst, сезон 3 эпизод 12) особенности диагностики болезни Фабри.
Болезнь Фабри
Болезнь Фабри – наследственное заболевание, при котором дефект в структуре генов обуславливает недостаточную активность или отсутствие фермента α-галактозидазы A, накопление в органах промежуточных продуктов липидного обмена. Симптоматика включает боли в конечностях, уменьшение потоотделения, депрессию, быструю утомляемость, почечную и сердечную недостаточность, острые нарушения мозгового кровообращения. Для диагностики используется исследование активности фермента и количества гликосфинголипидов в крови и тканях, секвенирование генетического материала. Лечение основано на ферментозаместительной терапии.
МКБ-10
Общие сведения
Болезнь Фабри получила свое название по фамилии немецкого врача-дерматолога Джона Фабри, который в 1898 году подробно описал симптомы патологии у мальчика 13 лет. В это же время схожий клинический случай был выявлен врачом-дерматологом из Великобритании Вильямом Андерсеном, поэтому заболевание имеет другое распространенное название – болезнь Андерсона-Фабри. Менее известные синонимы – церамидтригексозидоз, диффузная универсальная ангиокератома, наследственный дистонический липидоз. Эпидемиология зависит от расовой и этнической принадлежности, составляет 1 случай на 40-120 тыс. новорожденных. Наиболее высокая распространенность определяется в США.
Причины болезни Фабри
Заболевание обусловлено мутацией гена GLA, который кодирует структуру фермента альфа-галактозидазы А, участвующего в расщеплении гликосфинголипидов. Ген локализован на длинном плече Xq 22.1 хромосомы. Полипептидная структура фермента состоит из 429 остатков аминокислот, наличие и порядок расположения которых определяются 1290 парами оснований гена. В результате научных исследований выявлено 599 вариантов мутаций и полиморфизмов гена, которые изменяют стабильность и активность галактозидазы. Самые распространенные причинные мутации – миссенс и нонсенс, на их долю приходится до 93% случаев болезни.
Механизм наследования патологии определен как рецессивный X-сцепленный, но продолжает изучаться. У гемизиготных мужчин присутствует единственная мутантная X-хромосома. Такой набор представляет классический фенотип заболевания. Больные мужчины способны передать мутацию только дочерям, сыновья остаются здоровыми. Дочери, получившие измененные гены от родителя, являются гетерозиготными и, согласно закону рецессивного наследования, должны оставаться клинически здоровыми – носителями. Вероятность передачи патологического аллеля от матери детям обоих полов составляет 50%.
В ходе клинических исследований установлено, что у большинства женщин с рецессивным мутантным геном болезнь проявляется симптомами умеренной выраженности, имеет позднее начало и медленное прогрессирование. У части пациенток выявляются тяжелые проявления патологии, которые требуют неотложной медицинской помощи. Причины развития тяжелых форм заболевания у гетерозиготных женщин остаются неизвестными. Возможно существование феномена инактивации нормальной X-хромосомы.
Патогенез
Патогенетическая основа болезни – дефицит альфа-галактозы А. В норме этот фермент расщепляет терминальные гликолипиды а-галактозила. При ферментной недостаточности в клеточных лизосомах накапливаются промежуточные продукты обмена – тригексозилцерамид и дигалактозилцерамид. Содержание церамид-тригекосзида значительно увеличивается в эндотелиальных и гладкомышечных клетках сосудов, в эпителиальных и перителиальных клетках многих органов. Молекулы дигалактозил-церамида концентрируются в почках, камерах сердца, ЦНС.
Таким образом, основой патогенеза является нарушение метаболизма мембранных гликосфинголипидов. Их депонирование усиливается при повышенном уровне циркулирующих липидов, поступающих в мембраны методом активного всасывания и диффузии. Основные симптомы заболевания обусловлены скоплением продуктов обмена сфинголипидов в малых и крупных сосудах, включают боль в руках и ногах, приступы лихорадки, пурпурные кожные высыпания. Патологические процессы развиваются постепенно, полиморфная клиническая картина зачастую наблюдается у подростков и детей, у взрослых в большей степени поражается один орган.
Классификация
Поскольку патология является генетической, ее начало приходится еще на внутриутробный период. Из-за относительно медленного прогрессирования симптоматика проявляется гораздо позже – к 6-12 или даже к 30 годам. С учетом времени дебюта заболевания и характера поражения внутренних органов выделяют две формы болезни:
- Классическая. Начинается в детстве и раннем подростничестве. Проявляется полиорганным поражением.
- Атипичная. Симптомы дебютируют в зрелом возрасте. Часто наблюдается изолированное повреждение одного органа: почек, сердца, мозга. У представителей мужского пола признаки более выраженные.
- Женская. Данная форма выделяется в англоязычной литературе. Для нее характерно медленно прогрессирующее течение со слабовыраженной симптоматикой, отсутствие ведущего пораженного органа.
Симптомы болезни Фабри
Наиболее частый начальный симптом, который выявляется у 70-80% больных с классическим вариантом патологии – акропарестезии (невропатические боли). Они локализуются в дистальных отделах ступней и ладоней, отличаются высокой интенсивностью и продолжительностью, по характеру – жгучие, колющие, изнуряющие. Бывают хроническими и кризовыми. Хронические боли постоянные, но имеют среднюю интенсивность. Кризы Фабри длятся несколько часов или суток, представляют собой быстро нарастающие болевые приступы, сопровождающиеся гипертермией.
У значительной части больных наблюдается гипогидроз и ангидроз – снижение или полное отсутствие потоотделения. Пациенты плохо переносят жару, часто перегреваются (например, при кризовых болях). Ухудшается работоспособность, снижается толерантность к физическим нагрузкам. Занятия спортом и физический труд провоцируют усиление болей и перегрев тела. Характерный внешний признак болезни – ангиокератомы, мелкие безболезненные папулы красновато-фиолетового цвета, которые располагаются скоплениями на коже. Чаще всего поражается область губ, пальцы рук и ног, гениталии.
Сердечно-сосудистая симптоматика включает аритмии, артериальную гипертензию, гипертрофическую кардиомиопатию. Подростки страдают от приступов гипертензии. Взрослые пациенты испытывают болевые ощущения в области грудной клетки, головокружения, учащенное сердцебиение и одышку, падают в обмороки. Повреждение почек на начальной стадии протекает бессимптомно. При длительном течении болезни снижается фильтрационная и концентрационная функция почек, обращение за врачебной помощью часто происходит уже при хронической почечной недостаточности, требующей проведения гемодиализа.
Неврологические симптомы более свойственны взрослым. Типичны головные боли, головокружения. Скопление продуктов метаболизма липидов в сосудах становится причиной нарушения кровообращения в головном мозге. Возникают транзиторные ишемические атаки и инсульты. Офтальмологические расстройства имеют специфические особенности. Наиболее распространенными являются вортексная кератопатия у детей и помутнение хрусталика (двусторонняя передняя и радиальная задняя катаракта) у лиц старшего возраста. Возможно образование конъюнктивальных аневризм, отек сетчатки глаза или папиллоэдема, оптический неврит с выпадением полей зрения, центральными скотомами. Изменения слуха представлены звоном в ушах и ухудшением восприятия звуков. В психоэмоциональной сфере больных преобладают депрессия и тревога.
Осложнения
Нейропатические боли при болезни Фабри с трудом купируются обезболивающими препаратами, поэтому становятся причиной депрессии, низкой мотивации к учебе, профессиональной деятельности и иной социальной активности. В тяжелых случаях акропарестезии провоцируются минимальной физической нагрузкой или умственным переутомлением. В итоге дети и подростки переходят на домашнее обучение, что сопровождается сужением круга друзей, формированием ощущения изолированности. Пациенты находятся в группе риска по совершению суицидальных попыток. Почечные, сердечно-сосудистые и цереброваскулярные осложнения могут привести к инвалидизации, смерти.
Диагностика
При подозрении на болезнь Фабри обследование пациента проводится врачами нескольких специальностей: неврологом, кардиологом, дерматологом, генетиком. Диагноз устанавливают на основе типичных клинических проявлений, отягощенного семейного анамнеза, а также по данным лабораторной и инструментальной диагностики. Болезнь Фабри важно дифференцировать с коллагенозами, фибромиалгиями, болезнью Рейно. Специфические методы исследования включают:
- Анализ активности альфа-галактозидазы. Исследованию подвергается кровь (лейкоциты), сухие пятна крови, материал биопсии почек, культура кожных фибробластов. У мужчин активность фермента снижена. У женщин могут определяться показатели, соответствующие норме, нижней границе нормы или незначительно сниженные.
- Количественный анализ сфинголипидов. Выполняется исследование количества глоботриазилсфингозина в плазме крови, сухих пятнах крови. Тест широко применяется при скрининговых обследованиях. Высокие показатели с большой вероятностью указывают на наличие заболевания. Результаты используются не только для подтверждения диагноза, но и для контроля эффективности лечения.
- Секвенирование ДНК. У больных мужчин мутации диагностируются в гемизиготном наборе, у болеющих женщин или женщин-носителей – в гетерозиготном. Исследуется ген GLA. Обнаружение в его структуре мутационных изменений является самым точным способом диагностики болезни Фабри, особенно в отношении женщин.
- МРТ головного мозга. На снимках Т2 часто присутствует высокоинтенсивный сигнал в белом веществе теменной и фронтальной коры. На Т1-взвешенном изображении выявляется гиперинтенсивный сигнал в сером веществе глубоких структур. Болезнь характеризуется изолированным поражением заднего бугорка таламуса. Кроме того, выявляются сосудистые мальформации.
- ЭКГ, Эхо-КГ, МРТ сердца. Типична гипертрофия левого желудочка, на раннем этапе заболевания – укорочение интервала P-R, на позднем этапе – предсердно-желудочковая блокада. На МРТ визуализируется позднее попадание контраста на внутреннюю поверхность левого желудочка.
Лечение болезни Фабри
Единственным эффективным методом борьбы с заболеванием является ферментозаместительная терапия. С начала 2000-х годов лечение проводится рекомбинантными препаратами α-галактозы A. Своевременная медикаментозная терапия позволяет снизить выраженность невропатических болей, уменьшить выраженность гипертрофии левого сердечного желудочка, восстановить функциональность почек. Для скорейшего улучшения самочувствия больных применяются симптоматические средства. Парестезии и боли купируются антиконвульсантами, местными средствами с лидокаином.
При недостаточности функций почек и артериальной гипертонии назначаются ингибиторы АПФ, блокаторы АТ1-рецепторов, гемодиализ. Для профилактики тромбозов и инсультов используются антиагреганты, при тахикардии – антиаритмические препараты. Методы лечения продолжают разрабатываться. В настоящее время потенциально успешным считается направление генной инженерии. Предполагается, что вскоре станет возможным внедрение в клетки человеческого организма структурно правильного гена, задающего производство альфа субъединицы галактозидазы A.
Прогноз и профилактика
Своевременное начало терапии обеспечивает благоприятное течение болезни: симптомы купируются полностью либо остаются слабовыраженными, качество жизни пациентов повышается, женщины становятся способными зачать и выносить ребенка. Без лечения средняя продолжительность жизни мужчин составляет 40-60 лет, женщин – 40-70 лет. Профилактические меры заключаются в выявлении носительства мутации, медико-генетическом консультировании пар, в которых имеется партнер с подтвержденным диагнозом или отягощенным семейным анамнезом. При наступлении беременности таким парам необходимо проведение преимплантационной и пренатальной диагностики.
Источники:
http://vse-pro-geny.ru/ru_disease_3_Khvoroba-Fabri_%D0%91%D0%BE%D0%BB%D0%B5%D0%B7%D0%BD%D1%8C-%D0%A4%D0%B0%D0%B1%D1%80%D0%B8.html
http://www.krasotaimedicina.ru/diseases/genetic/Fabry
http://urohelp.guru/pochki/oslozhneniya/sindrom-fankoni.html

