
Группа заболеваний буллезного эпидермолиза
Что такое Группа заболеваний буллезного эпидермолиза –
Данная группа патологий, объединенная под общим названием заболеваний буллезного эпидермолиза, включает несколько различных форм болезни. Однако некоторые ученые склонны считать их совершенно самостоятельными заболеваниями.
Патогенез (что происходит?) во время Группы заболеваний буллезного эпидермолиза:
В основе всей группы описываемых заболеваний лежит возникновение на коже и на слизистых оболочках очагов поражений в виде пузырей. При этом воспалительные изменения отсутствуют, так как, несмотря на такие проявления, эти болезни не являются инфекционными. Пузыри никогда не возникают сами по себе, чаще всего их появлению предшествует какое-либо раздражающее воздействие на кожу, обычно в виде трения или травмы. Невозможно точно сказать, насколько распространенной среди детей является данная патология, однако известно, что она встречается среди населения всего земного шара.
Несмотря на большое количество различных форм патологии, все их возможно вписать в довольно простую короткую классификацию:
- простой буллезный эпидермолиз;
- дистрофический эпидермолиз, включающий в себя два основных подвида:
- гиперпластический;
- полидистрофический.
Симптомы Группы заболеваний буллезного эпидермолиза:
Простой буллезный эпидермолиз
Чаще всего заболевание развивается непосредственно с первых дней жизни ребенка. Однако в литературе описаны случаи появления симптоматики и в более позднем возрасте. На коже больного появляются пузыри с тонкими стенками. Размеры их различные – от горошины до грецкого ореха, несколько реже встречаются более крупные. Они заполнены водянистой жидкостью, при надавливании на них больной испытывает достаточно ощутимые болевые ощущения. Если пузырь вскрыть, то после излития из него жидкости боль стихает, довольно скоро наступает полное заживление. Вполне понятно, что в основном эти высыпания располагаются на коже в тех местах, которые постоянно подвергаются механическим воздействиям – локти, колени, лодыжки, кисти, стопы, ягодицы. Очередное обострение патологического процесса всегда происходит в летнее время года, после того, как больной принимает теплую ванну. Самостоятельное улучшение состояния отмечается зимой и во время периода полового созревания. Чаще поражаются мальчики. В основном проявления заболевания имеются только на кожных покровах, однако у части больных процесс может распространяться также и на слизистые оболочки, половые органы. На слизистых в местах травмирования образуются такие же пузыри, как и на коже, но они являются очень напряженными. После опорожнения пузыря образуется дефект слизистой оболочки, который быстро заживает. Рубцы не образуются никогда. Больной может лишь испытывать некоторое время чувство зуда и жжения в тех местах, где располагались пузыри. Ногти, зубы и волосы никогда не поражаются.
Летний буллезный эпидермолиз (синдром Вебера-Коккейна)
Летний буллезный эпидермолиз (синдром Вебера-Коккейна) – особая разновидность эпидермолиза. Начало приходится на период детства или юности. Пузыри на коже образуются после травмирования ее или длительного нахождения больного в условиях повышенной температуры. Провоцирующими факторами являются ходьба, теплая обувь, пребывание на курортах в странах с жарким климатом. Очаги поражения преимущественно располагаются в области кистей и стоп. Очень часто у таких больных обнаруживается повышенная потливость ладоней и подошв.
Дистрофический буллезный эпидермолиз
Как уже указывалось выше, данная патология может протекать в трех несколько отличных друг от друга формах – гиперпластической, альбопапулоидной и полидиспластической.
Гиперпластическая форма возникает сразу после рождения ребенка, во время периода грудного вскармливания или же несколько позже, но непременно в детском возрасте.Так же, как и при других разновидностях эпидермолиза, заболевание проявляется в виде накожных пузырей, которые возникают после имевшей место травмы. Чаще всего они расположены в области разгибательных поверхностей суставов на руках и ногах, в области кистей, стоп, ягодиц. Часто отмечается утолщение рогового слоя кожи на подошвах и кистях, а также утолщение ногтей, повышение потливости. У пятой части всех больных имеет место поражение слизистых оболочек, половых органов, конъюнктив. При таком расположении пузыри довольно быстро вскрываются, на их месте образуются дефекты, которые впоследствии плохо и долго заживают. Пузыри же на коже после вскрытия оставляют грубые рубцы, трудно поддающиеся хирургическому лечению. На слизистых же оболочках образуются рубцы с атрофией тканей. Часто из-за этого происходит деформация языка, в результате чего сильно нарушается речь больного, затрудненным становится прием пищи. Гиперпластическая разновидность заболевания часто сопровождается поражением зубов, избыточным оволосением, нарушениями формы и структуры ногтей. Часто у таких больных одновременно имеется ихтиоз. Несмотря на это, заболевание в целом протекает достаточно благоприятно, обострения могут возникать в летнее время года, особенно во время жары. Зимой к обострениям могут приводить различные состояния, связанные с воздействием на организм повышенной температуры, например прием горячих ванн, ношение слишком теплой одежды и обуви.
Альбопапулоидная форма эпидермолиза
Заболевание наследуется по аутосомно-доминантному принципу. Все носители патологического гена являются больными. Первые признаки возникают сразу после рождения ребенка или в течение первых двух месяцев жизни. Период наибольшего разгара симптоматики имеет место до 2-летнего возраста.
Кожные элементы в виде пузырей расположены чаще всего в области рук и ног, но иногда могут появляться и на туловище. Чаще всего это пояснично-крестцовая область, ягодицы, области надплечий. После заживления на местах пузырей остаются грубые рубцы и полости внутри кожи. Слизистые оболочки поражаются в очень небольшой степени, в основном во рту. Пузыри здесь возникают далеко не во всех случаях заболевания. Вместо пузырей тут чаще образуются очень мелкие пузырьки или такие же мелкие полости, которые при осмотре очень легко заметить, так как они имеют значительно более бледную окраску по сравнению с соседними областями. На местах всех этих элементов впоследствии остаются дефекты слизистой оболочки, которые довольно быстро совершенно бесследно исчезают. Имеются почти всегда нарушения со стороны ногтей рук и ног, которые в ряде случаев могут вообще отсутствовать. Выявляется повышенная сухость кожи, посинение в области кончиков пальцев, носа, ушей. При этом больной не предъявляет врачу совершенно никаких жалоб, так как общее состояние его практически никоим образом не страдает. С наступлением периода полового созревания пузыри у большей части больных самостоятельно исчезают, а на их местах остаются небольшие возвышения кожи бледного цвета. У некоторых детей выздоровление наблюдается еще раньше. В дальнейшем указанные образования начинают увеличиваться в размерах, приобретают цвет слоновой кости. Они сливаются между собой, в результате чего образуются очень большие бляшки на коже больного. Контуры их остаются довольно четкими, поэтому при осмотре больного их довольно легко выявить.
Полидиспластический буллезный эпидермолиз
Полидиспластический буллезный эпидермолиз является более редкой разновидностью заболевания, чем вышеперечисленные. Это одна из наиболее тяжело протекающих форм патологии. Начало ее развития приходится непосредственно на момент рождения ребенка.
В отличие от других форм эпидермолиза, пузыри при данной разновидности могут развиваться на коже не только в результате действия травмирующих факторов, но и сами по себе. Они наполнены кровянистой жидкостью, некоторые могут содержать жидкость прозрачного цвета. Пузыри располагаются не только в излюбленных для эпидермолиза местах (локти, колени, стопы, кисти), но и по всему телу больного. Почти всегда поражение захватывает также слизистые оболочки рта и пищевода, глотки. Именно здесь появляются очаги, которые возникают после первого прикладывания ребенка к груди матери. На слизистых оболочках появляются пузыри с характерным внешним видом, после вскрытия которых остаются очень грубые рубцы. Чаще всего они располагаются на языке, а оттуда переходят на щеки, что резко ограничивает открывание рта ребенком, нарушает процессы кормления. Зачастую в местах локализации рубцов на слизистых оболочках появляются очаги ороговения, которые в норме никогда не образуются. Иногда могут возникать многочисленные и мелкие узелки. Если развиваются все вышеуказанные элементы, то на их месте в дальнейшем образуются все новые пузыри, содержащие кровь и покрытые своеобразными пленками. Затем на этих местах остается налет, образующийся из остатков пузырей и этих пленок. После снятия налета шпателем обнаруживается болезненный кровоточащий дефект кожи, иногда даже в виде язвы. В свою очередь, возникающие на их местах рубцы еще больше способствуют нарушению подвижности языка ребенка. Формируется явление, называемое в медицинской практике порочным кругом. На слизистой оболочке щек возникают участки, имеющие значительно более бледный цвет по сравнению с окружающими. Патологический процесс практически всегда захватывает гортань, глотку и пищевод. В результате голос больного ребенка становится тихим, хриплым, осипшим. Очень большие дефекты в виде эрозий образуются на стенках пищевода и иногда даже желудка, в результате чего питание ребенка становится резко затрудненным, развивается истощение, доходящее порой до крайней степени выраженности. Возможно появление сквозных отверстий в стенках указанных органов, на фоне которых формируются грозные для жизни осложнения. Встречаются поражения заболеванием прямой кишки и ануса, в итоге отхождение стула может стать совершенно невозможным. Если же поражаются слизистые оболочки половых органов и мочевыделительной системы, то это в дальнейшем ведет к нарушению нормального оттока мочи, осложнениям со стороны вышеуказанных систем. При поражениях пальцев сильно страдают ногти, иногда они полностью исчезают либо деформируются. Встречаются также сращения пальцев на руках и ногах, которые не являются пороками развития, а формируются в результате непосредственно заболевания, во время жизни ребенка. Руки в локтевых суставах и ноги в коленных могут плотно фиксироваться в сгибательном положении, движения в них значительно затрудняются или полностью утрачиваются. Все вышеперечисленные нарушения встречаются наиболее часто и у большинства больных детей. Однако круг расстройств описываемой разновидности эпидермолиза может быть намного шире. Часто встречаются самые различные виды поражения волос, ногтей, зубов, скелета, суставов. Как правило, такие дети имеют низкий рост и массу тела, у них отмечаются значительные нарушения со стороны иммунных сил организма. Всегда указанная патология приводит к развитию инвалидности. Не исключены и случаи смертельных исходов. Часто развивается малокровие, в результате потери большого количества крови из дефектов, возникающих вследствие вскрытия пузырей на слизистых оболочках. Если не уделять достаточного внимания уходу за кожей и слизистыми ребенка, то на местах рубцов возникают опухолевые процессы, в ряде случаев даже различные формы рака.
Однако не всегда заболевание протекает настолько тяжело. Встречается также его местная форма, когда поражения на слизистых ограничиваются лишь одним или несколькими небольшими участками. Течение патологии в этих случаях намного более благоприятное. При этом на коже пузыри в основном возникают в области кистей, стоп, локтей и коленных суставов. Грубых больших рубцов не бывает никогда. Наблюдается незначительная атрофия кожи, образование в ней небольшого количества кистозных полостей. Слизистые оболочки страдают еще меньше. Небольшие изменения выявляются со стороны ногтей.
Лечение Группы заболеваний буллезного эпидермолиза:
Так как эпидермолизы относят к обменным наследственным заболеваниям, то возможности по их лечению несколько шире. Известно, что в основе этой группы патологий лежит нарушение в обмене особого белка, регулирующего чувствительность различных тканей, в том числе и кожи, к действию температур. При этом имеет место нарушение обменных процессов во всех клетках. Поэтому наиболее действенным методом медикаментозной терапии является назначение больному анаболических стероидов.
Применяются витамины, в частности Е, гормоны коры надпочечников с целью повышения общей устойчивости организма к действию различных неблагоприятных факторов. Назначаются аминокислоты, биологически активные вещества, другие препараты, стимулирующие обменные процессы.
Также с целью общеукрепляющего эффекта можно применять лекарственные вещества, содержащие кальций, железо, цинк, серу, алоэ, взвесь человеческой плаценты. Применяются иммуноглобулины, представляющие собой набор необходимых для организма ребенка антител. При дисбактериозе назначают про- и эубиотики. Особенно важным препаратом является аскорбиновая кислота.
На коже необходимо своевременно вскрывать появившиеся пузыри, на эти места применяются в дальнейшем дезинфицирующие растворы и мази, способствующие ускорению процессов заживления. Нужен тщательный правильный уход за кожей больного, нельзя допускать ее травмирования и действия на нее больших температур. При присоединении инфекционных осложнений назначаются антибиотики.
Меры профилактики и реабилитации такие же, как и у больных с другими наследственным заболеваниями кожи.
Прогноз
Различный, в зависимости от степени тяжести заболевания. При местных формах благоприятный во всех отношениях. При тяжелом течении возможны смертельные исходы. Абсолютное большинство таких детей в последующем остаются инвалидами на всю жизнь.
К каким докторам следует обращаться если у Вас Группа заболеваний буллезного эпидермолиза:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Группы заболеваний буллезного эпидермолиза, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Буллезный эпидермолиз
Буллезный эпидермолиз – группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий – «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
МКБ-10
Общие сведения
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Причины буллезного эпидермолиза
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация буллезного эпидермолиза
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
- Простой буллезный эпидермолиз – имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера-Коккейна, Кёбнера, Доулинга-Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
- Пограничный буллезный эпидермолиз – делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
- Дистрофический буллезный эпидермолиз – имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
- Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи – эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе – киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Симптомы буллезного эпидермолиза
Проявления буллезного эпидермолиза разных типов объединяет одно – развитие пузырей и эрозий в ответ на механическое воздействие на кожу. Различается лишь степень выраженности этих изменений, локализация, время существования и результаты заживления. При локализованной форме простого буллезного эпидермолиза (подтип Вебера-Коккейна) поражения располагаются только на определенном участке тела (руки, стопы). В младенческом возрасте возможна более широкая площадь появления пузырей, но с возрастом их выраженность уменьшается. Напротив, генерализованный подтип Доулинга-Меары характеризуется развитием мелких везикулярных высыпаний на значительной площади тела. Такой тип буллезного эпидермолиза возникает с самого раннего детства и может стать причиной смерти ребенка, итогом разрешения пузырьков может быть гиперкератоз, нарушения пигментации кожи, иногда возникает поражение слизистых.
Пограничная форма буллезного эпидермолиза протекает намного более тяжело, особенно так называемый летальный подтип Херлитца. При этом наблюдается повышенная ломкость кожных покровов, образование большого количества пузырьков, эрозий, на лице и спине часто возникают симметричные грануляции. Поражаются и слизистые оболочки рта, обнаруживается гипоплазия эмали и обусловленный ею тяжелый кариес. Столь тяжелое течение пограничного буллезного эпидермолиза часто становится причиной летального исхода в первые годы жизни. У выживших больных во взрослом возрасте формируются контрактуры суставов, поражение почек, потеря ногтей. Более легкая атрофическая форма пограничного буллезного эпидермолиза также характеризуется обширными высыпаниями, после разрешения которых формируются атрофические участки и рубцы. Также она часто приводит к дистрофии ногтей и рубцовой алопеции.
Дистрофический буллезный эпидермолиз практически всегда является генерализованным и поражает обширные участки тела. Доминантный вариант заболевания в целом отличается более доброкачественным течением, образование пузырей и их разрешение происходит медленно, однако большинство больных в конце концов теряют ногти на руках. После заживления эрозий на поверхности кожи формируются заметные рубцы. Рецессивный вариант дистрофического буллезного эпидермолиза, особенно его тяжелый генерализованный подтип, протекает намного тяжелее: помимо высыпаний у больных часто регистрируются псевдосиндактилии, обширные шрамы, потеря ногтей. Возникает поражение костей скелета, на месте заживших шрамов с годами может развиваться плоскоклеточный рак. Проблемой является еще и высокая устойчивость подтипа Аллопо-Сименса к терапевтическим мероприятиям.
Осложнения любого типа буллезного эпидермолиза сводятся к риску развития шока (при обширных поражениях), присоединения вторичной инфекции и спровоцированного ею сепсиса, обезвоживания больных. В большинстве случаев терапевтические процедуры производят только с целью недопущения этих состояний. Вероятность развития осложнений тем выше, чем большую область тела занимают патологические очаги и чем деструктивнее их характер (напряженные пузыри, эрозии, язвы).
Диагностика буллезного эпидермолиза
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода. Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение буллезного эпидермолиза
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз буллезного эпидермолиза
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Помочь тем, кого нельзя обнять
Обзор
Автор
Редактор
Статья на конкурс «био/мол/текст»: «Мальчик, который остался без кожи». Это не название очередного фильма ужасов. Это документальный фильм режиссера Патрика Коллертона, запечатлевшего последние месяцы жизни 36-летнего Джонни Кеннеди, страдавшего редким генетическим заболеванием — дистрофическим буллезным эпидермолизом. Фильм привлек внимание пяти миллионов зрителей по всей Великобритании и помог собрать 500 000 фунтов стерлингов на благотворительность для международной ассоциации DEBRA, чья деятельность направлена на изучение и лечение буллезного эпидермолиза. Жизнь людей с этим диагнозом и их родных — это путь, который осилит не каждый: он тяжел как физически, так и морально, и требует мужества и невероятной воли к жизни. Детей, страдающих этим недугом, называют детьми-бабочками, сравнивая хрупкость их кожи с хрупкостью крыла бабочки. Что же такое буллезный эпидермолиз и каковы прогнозы на будущее?
Обратите внимание!
Эта работа опубликована в номинации «лучшая обзорная статья» конкурса «био/мол/текст»-2015.
Спонсором номинации «Лучшая статья о механизмах старения и долголетия» является фонд «Наука за продление жизни». Спонсором приза зрительских симпатий выступила фирма Helicon.
Спонсоры конкурса: Лаборатория биотехнологических исследований 3D Bioprinting Solutions и Студия научной графики, анимации и моделирования Visual Science.
Я знаю только один способ быть в ладу с собственной совестью: этот способ — не уклоняться от страдания.
Антуан де Сент-Экзюпери
Что такое буллезный эпидермолиз?
Буллезный эпидермолиз (БЭ) — группа редких генетически и клинически гетерогенных заболеваний, характеризующихся образованием пузырей и эрозий на коже и слизистых оболочках в результате незначительной механической травмы [1].
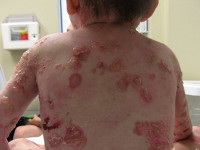
Кожа человека состоит из трех слоев: наружного — эпидермиса, среднего — дермы — и самого нижнего — подкожно-жировой клетчатки (гиподермы) (рис. 1).

Рисунок 1. Строение кожи человека. Рисунок с сайта lechenie-simptomy.ru.
Эпидермис кожи человека состоит из пяти различных слоев эпителиальных клеток, или кератиноцитов, самый нижний из которых — базальный эпителий — прикрепляется к дерме с помощью множества разных белков (рис. 2). Эти белки не только определяют стабильность соединения клеток базального эпителия с дермой, но и их собственную прочность. Мутации в генах, кодирующих эти белки, приводят к снижению прочности этого соединения, делая кожу чрезвычайно чувствительной к механическим воздействиям. Даже самое легкое надавливание — трение швов одежды, нежные объятия матери — может привести к образованию пузырей и причинить нестерпимую боль (рис. 3) [1].

Рисунок 2. Белки, участвующие в патогенезе различных типов наследственного БЭ. ПБЭ — простой БЭ; ПогрБЭ — пограничный БЭ; ДБЭ — дистрофический БЭ; СК — синдром Киндлера; БМ — базальная мембрана. Рисунок из [1], с изменениями.

Рисунок 3. Микроструктура образца кожи больного БЭ. При БЭ соединение между эпидермисом и дермой непрочное. В результате механического воздействия эпидермис отслаивается от дермы. В месте повреждения увеличивается проницаемость капилляров, и жидкая часть крови (плазма), а также клетки, участвующие в воспалении (лейкоциты), выходят в рану — образуется воспалительный экссудат. В месте отслоения формируется пузырь, который увеличится в размерах, если его вовремя не проткнуть стерильной иглой. Рисунок с сайта dermaamin.com, с изменениями.
Как правило, заболевание заметно сразу после рождения ребенка или в первые месяцы его жизни. Каждый новый день — настоящее испытание для больных БЭ и их опекунов: смена повязок, защищающих хрупкую кожу от повреждений и закрывающих уже существующие раны, обработка ран антисептиками — эти жизненно важные, чрезвычайно болезненные и изнурительные процедуры являются практически единственной составляющей современного лечения БЭ. При тяжелых формах заболевание поражает помимо кожных покровов слизистые оболочки и даже внутренние органы. БЭ относится к так называемым орфанным, то есть редким, заболеваниям. Частота встречаемости различных типов БЭ варьирует от 1:30 000 до 1:1 000 000 и зависит от популяции [2].
Классификация
В зависимости от уровня, на котором происходит повреждение кожи, выделяют четыре типа БЭ: простой, пограничный, дистрофический и синдром Киндлера.
Простой БЭ относится к так называемому интраэпидермальному типу БЭ, то есть повреждения затрагивают только эпидермис. Простой БЭ развивается в результате мутаций в генах белков, обеспечивающих прочность клеток базального слоя эпителия (рис. 2). Пузыри образуются ближе к поверхности кожи. Это самая легкая форма БЭ, при которой происходит полноценное заживление ран. Пограничный и дистрофический БЭ относятся к субэпидермальным типам БЭ, поскольку затрагивают не только эпидермис, но и дерму. При этих типах БЭ поврежденными оказываются белки, участвующие непосредственно в соединении двух слоев кожи (рис. 4). Полноценного заживления ран не происходит — на месте нормальной кожи в конечном счете образуются рубцы. Пограничный БЭ, в свою очередь, подразделяется на два типа: Герлиц и не-Герлиц, первый из которых летален. Дистрофический же БЭ в зависимости от типа наследования подразделяется на доминантную и рецессивную формы. Синдром Киндлера относится к смешанному типу БЭ, поскольку пузыри образуются на разных уровнях [1].

Рисунок 4. Иммунофлуоресцентное картирование образца кожи пациента с рецессивным дистрофическим БЭ. Для картирования использовали антитела к коллагену VII (зеленый цвет). а — Пониженное количество коллагена VII в биоптате от пациента с рецессивным дистрофическим БЭ. б — Кожа здорового донора. Ядра клеток окрашены йодидом пропидия (красный цвет). Рисунок из [3], с изменениями.
Диагностика
Для того чтобы подтвердить диагноз БЭ, необходимо провести тщательное обследование пациента. Самым важным этапом диагностики является исследование биоптата поврежденных участков кожи. Для этого используют такие методы, как световая, флуоресцентная и электронная микроскопия, а также генетический анализ.
Исследование с помощью световой микроскопии позволяет отличить интраэпидермальный тип БЭ от субэпидермальных. Для более точного определения уровня повреждения используют электронную и флуоресцентную микроскопии. С помощью электронной микроскопии можно отличить рецессивную форму дистрофического БЭ от доминантной (рис. 5). Так, при рецессивном дистрофическом БЭ наблюдается значительное снижение или полное отсутствие продукции коллагена VII типа, в то время как при доминантном дистрофическом БЭ дефектный белок вырабатывается в нормальном или слегка сниженном количестве. Однако из-за трудоемкости методов электронной микроскопии в настоящее время на первый план выходит микроскопия флуоресцентная. С помощью окрашивания флуоресцентными антителами этот метод позволяет визуализировать белки, участвующие в патогенезе заболевания, а также оценить их продукцию и распределение (рис. 4) [3].

Рисунок 5. Ультраструктура образца поврежденного участка кожи пациента с рецессивным дистрофическим БЭ. Звездочками обозначены места разрывов, расположенные сразу под темной пластинкой базальной мембраны (бесклеточного слоя, отделяющего дерму от эпидермиса), на которую указывают «галочки». Стрелками обозначены участки с единичными фибриллами коллагена VII. Изображение получено методом электронной микроскопии. Рисунок из [3].
Но только генетический анализ (ДНК-диагностику) можно назвать оптимальным методом для определения типа наследования и специфических мутаций, имеющихся у больных БЭ, а также наиболее точным методом различения клинических форм простого, пограничного и дистрофического БЭ [4].
Существует также пренатальная (до рождения) диагностика БЭ, материалом для которой служит ДНК из околоплодной жидкости, забираемая в первом триместре беременности (до 11 недель) [4].
Лечение
К сожалению, в настоящее время БЭ неизлечим. Основная терапия направлена на предотвращение образования новых пузырей и эрозий, лечение ран и предотвращение их инфицирования. Главные задачи лечения — защита хрупкой кожи пациентов от механических воздействий (использование специальных повязок) и обработка уже существующих ран (применение антисептиков, наложение повязок).
Но наука не стоит на месте, и ученые всего мира отчаянно ищут лекарство от страшного недуга. Наиболее проработаны три перспективных вида терапии БЭ: протеиновая, клеточная и генная. Рассмотрим эти подходы подробнее.
Протеиновая терапия
При этом подходе в организм пациента вводится достаточное количество нормального белка. Исследования показали, что внутридермальное введение мышам с рецессивным дистрофическим БЭ очищенного человеческого коллагена VII приводит к формированию нормальных коллагеновых фибрилл в зоне соединения эпидермиса с дермой. При внутривенном введении очищенного белка наблюдается системное отложение коллагена VII в коже. Также было показано, что подобная терапия не только улучшает функциональное состояние кожи, но и способствует заживлению ран. Основываясь на результатах доклинических исследований, компания Lotus Tissue Repair, впоследствии вошедшая в состав Shire Pharmaceuticals, начала проведение расширенных доклинических исследований внутридермальных и внутривенных инъекций коллагена VII на модели рецессивного дистрофического БЭ у собак, а также первую стадию клинических исследований [3, 5].
Клеточная терапия
При этом способе лечения в организм пациента вводится необходимое и достаточное количество клеток, содержащих нормальный ген, кодирующий нужный белок.
Одним из перспективных видов клеточной терапии является внутридермальная инъекция аллогенных (полученных от здоровых доноров) фибробластов. Так, было показано, что подобный подход восстанавливает синтез коллагена VII и стабилизирует соединение эпидермиса с дермой в мышиной модели рецессивного дистрофического БЭ. Следующий шаг на пути к реализации этого вида терапии уже предпринят: нескольким пациентам с рецессивным дистрофическим БЭ были введены аллогенные фибробласты. И хотя спустя две недели после инъекции введенные клетки не обнаруживались, уровень продукции коллагена оставался повышенным как через две недели, так и через три месяца. Ученые считают, что основной терапевтический эффект инъекции аллогенных фибробластов заключается в увеличении продукции эпидермального фактора роста HB-EGF, а также в увеличении экспрессии гена коллагена VII в кератиноцитах и фибробластах реципиента. Однако механизм этого явления пока не установлен [3, 6].
Аллогенная трансплантация костного мозга также может быть перспективным способом лечения БЭ. Так, на мышиной модели рецессивного дистрофического БЭ было показано, что трансплантация костного мозга приводит к облегчению симптомов болезни. Вслед за обнадеживающими результатами были начаты клинические испытания подобного метода терапии. Однако пока результаты неутешительны: пять из двадцати пациентов умерли от прогрессии заболевания или осложнений после трансплантации [3, 6].
Генная терапия
Наконец, последний и, пожалуй, самый перспективный подход — генная терапия (рис. 6). В этом случае пациентам пересаживаются аутологичные (сделанные из собственных клеток) трансплантаты, в клетках которых с помощью методов генетического редактирования дефектный ген заменяется нормальным. И хотя подобная процедура не приводит к полному исцелению, она способна временно устранить симптомы БЭ, а также является относительно простой в техническом отношении.

Рисунок 6. Редактирование собственной ДНК в лечении БЭ. а — Принцип генной терапии. На первом этапе культивируют эпидермальные стволовые клетки из биоптатов кожи пациента с БЭ. Затем стволовые клетки подвергают процедуре генетического редактирования, в результате которой дефектный ген замещается нормальным. После того как пласт из генетически отредактированных кератиноцитов будет проверен на безопасность, его трансплантируют пациентам. б — Трансплантаты, сформировавшиеся из «правильных» кератиноцитов пациента с рецессивным дистрофическим БЭ. Рисунок из [7].
Первые успехи генной терапии были достигнуты в Италии в 2006 году, когда в эпидермальные стволовые клетки пациента с пограничным БЭ с помощью ретровирусной конструкции был доставлен нормальный ген LAMB3, кодирующий одну из субъединиц белка ламинина-332. Из «исправленных» клеток были созданы эпидермальные трансплантаты, которые затем были пересажены на ноги пациента. В результате из них образовалась полноценная в функциональном отношении кожа. Исследование показало, что выживание даже небольшого числа стволовых клеток в подобных трансплантатах приводит к успешному восстановлению нормальных функций кожи. Спустя восемь лет продукция целевого белка LAMB3 в коже пациентов всё еще сохранялась. Не было обнаружено ни пузырей, ни признаков воспаления, опухолевого роста или какого-либо иммунного ответа в области трансплантата. В июле 2014 года в Австрии провели вторую подобную операцию. Профессор Альфред Лэйн из Стэнфордского университета (Калифорния) вместе с сотрудниками начал клинические испытания подобной технологии, нацеленной на восстановление синтеза коллагена VII в эпидермальных стволовых клетках пациента с рецессивным дистрофическим БЭ. Спустя 30 дней после операции в областях трансплантации не было отмечено никаких отклонений, а уровень продукции коллагена VII был гораздо выше исходного [7].
Конечно, и в случае генной терапии существует множество сложностей. Так, замена гена с использованием вирусных конструкций и невирусных систем редактирования ДНК (ZFN, TALEN и CRISPR/Cas9)* связана с риском встраивания гена не в то место генома, которое являлось мишенью. Оценить последствия такого встраивания довольно сложно. Поэтому такой подход требует большой осторожности.
* — Методы ZFN, TALEN и CRISPR/Cas9 основаны на сайт-специфическом действии нуклеаз in vivo. Эти системы обычно состоят из двух модулей, один из которых распознает целевую олигонуклеотидную последовательность, а другой режет цепи ДНК: «А не замахнуться ли нам на. изменение генома?» [8], «CRISPR-системы: иммунизация прокариот» [9], «Мутагенная цепная реакция: редактирование геномов на грани фантастики» [10]. — Ред.
Другая проблема подобной терапии — возможный риск развития иммунного ответа на экспрессируемый новый белок. Особенно велик риск для тех пациентов, чьи клетки несли мутации, полностью исключающие синтез определенного белка. В то же время известно, что у некоторых пациентов с БЭ есть участки кожи, на которых не образуются пузыри и продуцируется повышенный уровень белка. Это явление названо обратным мозаицизмом, или естественной генной терапией, и может предотвращать развитие иммунной реакции на синтезируемый в результате генной терапии белок. Более того, такие «самоизлечившиеся» клетки можно использовать для клеточной терапии, и тогда потребность в генной коррекции исчезнет. Однако до сих пор попытки создать трансплантаты на основе таких клеток у больных с пограничным БЭ не увенчались успехом из-за малого числа подобных клеток в пересаживаемом материале [11].
Заключение
С тех пор как в 1886 году немецкий врач Кебнер впервые употребил термин «буллезный эпидермолиз», многое стало известно о патогенезе этого заболевания. Тем не менее до сих пор основным инструментом терапии является правильный уход за такими больными. Всё дело в том, что разработка и производство лекарств для лечения орфанных заболеваний представляет собой наукоемкий и дорогостоящий процесс, увы, невыгодный с коммерческой точки зрения из-за малого потребительского рынка. К сожалению, государство не в состоянии самостоятельно обеспечить решение этой проблемы. В 1978 году в Великобритании была основана ассоциация DEBRA, в настоящее время насчитывающая более 40 стран-участниц. Деятельность ассоциации направлена в первую очередь на медицинскую и социальную помощь пациентам с БЭ и их родным. Помимо этого, ассоциация финансирует исследования в области БЭ и занимается привлечением общественного внимания к данной проблеме. Представителем ассоциации в России является фонд «Б.Э.Л.А. Дети-бабочки». Именно благодаря совместным усилиям государства и ассоциации DEBRA стало возможным проведение масштабных исследований БЭ. В результате многолетней работы ученых всего мира появилось несколько перспективных подходов к терапии БЭ, а некоторые из них сейчас проходят первую фазу клинических испытаний. И хотя впереди еще несколько этапов тщательных исследований, первые положительные результаты дают надежду пациентам с этим редким генетическим заболеванием.
Больно, незаразно, не лечится и еще 9 фактов о буллезном эпидермолизе

Буллезный эпидермолиз — редкое генетическое заболевание кожи, о котором зачастую не знают не только обычные люди, но и врачи. В начале Недели буллезного эпидермолиза предлагаем полный ликбез по этому заболеванию
Буллезный эпидермолиз — это пожизненная проблема, от которой нельзя избавиться по желанию, с помощью дорогостоящего лечения или иностранных врачей. Поэтому буллезный эпидермолиз оказывает на больного сильное психологическое воздействие: наличие видимых изменений кожи и частое посещение больниц могут повлиять на социальную, учебную и профессиональную жизнь человека, значительно снизив ее качество. Сказывается и не всегда корректная реакция незнакомых людей.
Кроме того, несмотря на развитость информационных технологий, далеко не все врачи в России сегодня знают о буллезном эпидермолизе и могут поставить правильный диагноз. В итоге многих «бабочек» (так называют пациентов с диагнозом) годами «лечат» от мнимой пищевой аллергии или других заболеваний, не имеющих к реальности никакого отношения.
Чем больше общественность, в том числе и профессиональная, будет знать о буллезном эпидермолизе и связанных с ним проблемах, тем больше внимания, принятия и квалифицированной медицинской помощи будут получать такие больные. В России Неделю буллезного эпидермолиза проводит Благотворительный фонд «Дети-бабочки».

Маргарита Гехт,
ведущий врач-дерматолог благотворительного фонда «Дети-бабочки»
Что такое буллезный эпидермолиз
Буллезный эпидермолиз (БЭ) — не одно, а целая группа редких генетических заболеваний. У людей с таким диагнозом кожа и слизистые оболочки очень хрупкие. От малейшего прикосновения на них образуются пузыри. Лопаясь, они оставляют после себя болезненные раны.
Эти пузыри наверняка знакомы и вам. Такая влажная мозоль возникает на коже после целого дня, проведенного в тесных туфлях. Вечером вы нередко обнаруживаете ее уже вскрывшейся. Но если у обычных людей такие пузыри вздуваются из-за длительного трения тесной обуви и плотно прилегающей кожи, то у людей с БЭ — спонтанно или в результате малейших травм. Именно поэтому пациентов с этим заболеванием метафорично называют «бабочками». Как известно, даже легкое прикосновение к крылу бабочки снимает с него защитный слой, в результате чего она уже не может летать. При БЭ поврежденная кожа болит и может инфицироваться.
Чтобы понять, как возникает буллезный эпидермолиз, необходимо знать строение человеческой кожи.
Как устроена кожа
Кожа — самый большой орган нашего тела. Она защищает организм от микробов и аллергенов, помогает регулировать температуру тела и позволяет ощущать прикосновение, тепло и холод.
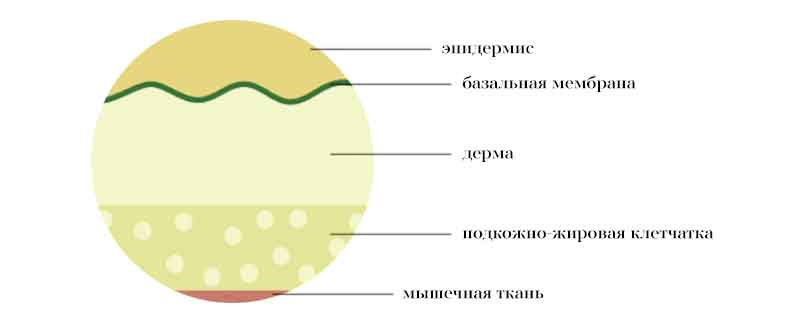
Кожа имеет три слоя:
Эпидермис
Внешний слой кожи, видимый глазу. Его основные функции — защитная и сохранения водонепроницаемого барьера — гидролипидной мантии.
Дерма
Этот слой находится под эпидермисом. Содержит плотную соединительную ткань, волосяные, сальные фолликулы, потовые железы, коллаген, эластин. В дерме проходят кровеносные сосуды и нервные окончания.
Между эпидермисом и дермой находится базальная мембрана — важная структура, соединяющая эти два слоя.
Гиподерма, или подкожно-жировая клетчатка
Глубокий слой кожи. Он состоит из жировой и соединительной ткани.
Работу, а главное, взаимодействие слоев кожи между собой определяют различные «помощники»: гены, ферменты, белки, дополнительные структуры и клетки. В случае нарушения работы хотя бы одного из «помощников» в коже происходят различные изменения. Так, при внезапно возникающих или вызванных искусственно стойких изменениях наследственных структур появляются генодерматозы, в том числе и буллезный эпидермолиз.
Что такое генодерматозы
Генодерматозы — наследственные заболевания кожи, насчитывающие несколько сотен нозологических форм и проявляющиеся различными патологическими процессами.
Причина их возникновения кроется в генах человека. Они хранят информацию, которая переводится в структуры кожи, содержащие белки, а именно — в кератин, коллаген, ламинин и интегрин, которые обеспечивают целостность и устойчивость кожи. При генетических поломках эти структуры перестают выполнять свою скрепляющую функцию, в результате кожа теряет целостность и становится хрупкой.
Буллезный эпидермолиз — один из самых тяжелых видов дерматозов данной группы.
Какие бывают виды буллезного эпидермолиза
Существует четыре основных формы буллезного эпидермолиза:
- простая;
- пограничная;
- дистрофическая;
- синдром Киндлера;
Классификация БЭ зависит от того, на каком уровне кожи произошел патологический процесс.
Простая форма
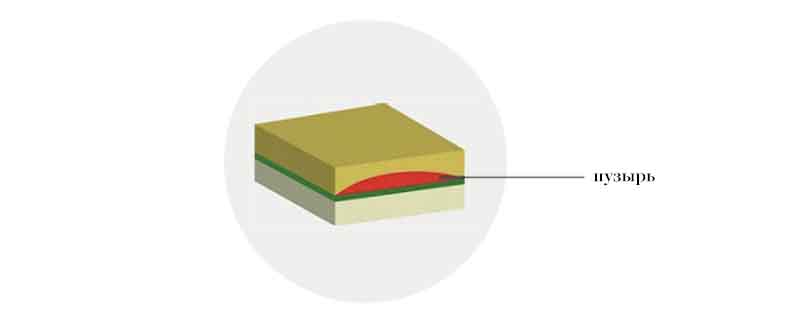
При данной форме БЭ пузыри появляются в пределах эпидермиса. Они не приводят к образованию рубцов, но причиняют сильную боль. Кроме того, летом, когда усиленное потоотделение провоцирует образование новых пузырей, простая форма БЭ всегда протекает тяжелее.
Пограничная форма
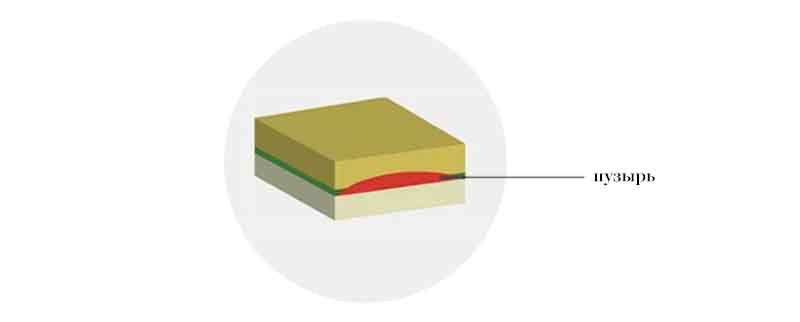
В данном случае расщепление происходит внутри базальной мембраны — структуры, которая «скрепляет» эпидермис и дерму. Более того, при определенном подтипе такой формы базальная мембрана вообще отсутствует. Из-за этого одна кожная структура может «находить» на другую, в результате чего на коже возникают многочисленные пузыри и обширные раны. Сливаясь, раны часто не оставляют на коже живого места и могут вызвать сепсис, а значит, потенциально опасны для жизни.
Дистрофическая форма
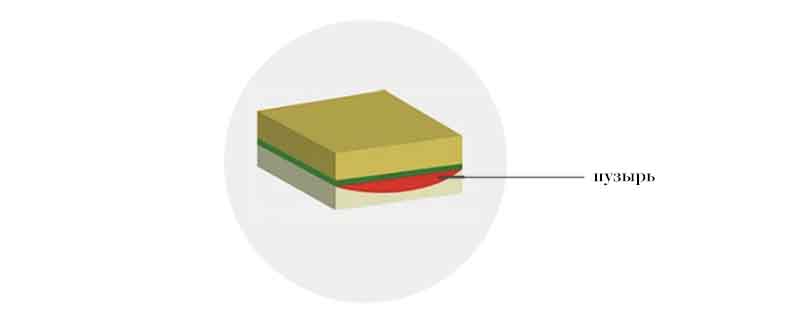
При данном типе БЭ расщепление происходит ниже базальной мембраны, в дерме. В этой области залегают ключевые структуры, которые определяют плотность и упругость кожи, — коллаген и эластин. При такой форме БЭ патологический процесс протекает именно в коллагене, в результате чего пузыри заживают с последующим образованием рубцовой ткани.
Синдром Киндлера
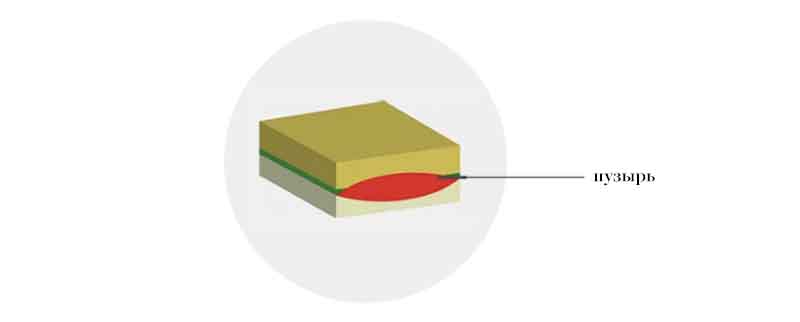
В этом случае пузыри могут образовываться одновременно на разных уровнях кожи. Кожные проявления могут быть вариабельны и зависят от степени вовлеченности гена в мутацию.
Чем отличаются врожденный и приобретенный БЭ
БЭ можно разделить на две группы — врожденный и приобретенный.
Врожденный БЭ, которому посвящена большая часть данной статьи, обусловлен генетическим дефектом. Этот дефект возникает еще внутриутробно, а сразу после рождения становятся видны внешние клинические проявления — раны на коже. Сегодня эта форма БЭ не имеет патогенетического лечения.
Приобретенный БЭ — это аутоиммунный дерматоз, связанный с выработкой специфических антител к собственному коллагену VII типа — наиболее важному компоненту фибрилл. Фибриллы — особые структуры в форме якоря, которые вместе с другими компонентами кожи скрепляют ее слои между собой.
При приобретенном БЭ образование пузырей происходит после связывания аутоантител — это антитела, которые выработались на собственные белки в результате воздействия на организм триггерных факторов (среди которых можно выделить стресс, перенесенные заболевания и оперативные вмешательства с коллагеном VII типа). Это, в свою очередь, приводит к активации каскада воспалительных реакций со стороны кожи.
Клинически заболевание характеризуется натянутыми пузырями на травмированных участках тела, которые заживают с образованием рубца. Так проявляется механобуллезная форма приобретенного БЭ. Второй наиболее частый подтип приобретенного БЭ — буллезное пемфигоидоподобное заболевание. Для него характерен кожный зуд и поражение слизистых оболочек. Для лечения приобретенного БЭ назначают местное и системное использование глюкокортикоидов и введение внутривенных иммуноглобулинов.
Как диагностируется буллезный эпидермолиз
Детальная лабораторная диагностика необходима для определения формы и подтипа БЭ и точной причины его возникновения на генетическом и белковом уровнях. Это помогает прогнозировать тяжесть состояния, оказывать надлежащую медицинскую помощь и участвовать в клинических испытаниях.
В лабораторной диагностике БЭ используются два основных метода:
1. Генетическое тестирование, которое направлено на выявление в ДНК мутации в конкретном гене, вызывающей заболевание.
2. Анализ образцов кожи с использованием методов, работающих на уровне белка, который позволяет выявить изменения в белке и компонентах кожи.
Влияет ли буллезный эпидермолиз только на кожу
Хотя физические эффекты БЭ затрагивают в основном кожу, при некоторых формах БЭ поражается не только кожа, но и слизистые оболочки внешних и внутренних органов: глаз, полости рта, пищевода, кишечника.
Заразен ли буллезный эпидермолиз
БЭ — генетическое заболевание. Это значит, что с ним рождаются. Заразиться буллезным эпидермолизом нельзя.
Как наследуется это заболевание
БЭ может быть унаследован одним из трех способов:
- аутосомно-доминантным наследованием;
- аутосомно-рецессивным наследованием;
- наследованием de novo.
Аутосомно-доминантное наследование
В этом случае один из родителей страдает заболеванием и имеет дефектный ген. Он передает измененный ген своему ребенку. Вероятность того, что любой их ребенок родится с БЭ, составляет 50%.
Аутосомно-рецессивное наследование
В этой ситуации оба родителя клинически здоровы и не имеют внешних проявлений заболевания, но являются его генетическими носителями. Чтобы их ребенок родился с БЭ, он должен унаследовать дефектный ген — часть гена от матери, часть — от отца. Возникает всем известная математическая комбинация, когда минус на минус дает плюс в виде болезни у ребенка. Вероятность того, что это произойдет, составляет 25%.
Мутации, вызывающие БЭ, которые происходят de novo
Человек, несущий вариант de novo, то есть родившийся с БЭ при здоровых родителях, в том числе не имеющих генных мутаций, имеет 50%-й шанс передать заболевание своим детям.

Можно ли вылечить БЭ
На сегодняшний день нет эффективного лечения ни одной из форм БЭ. Все доступное лечение направлено на облегчение симптомов, чтобы, в конечном счете, улучшить качество жизни человека с данным генодерматозом.
Какова продолжительность жизни пациентов с БЭ
Продолжительность жизни при буллезном эпидермолизе зависит от формы заболевания, качества ухода за кожей, профилактики внекожных проявлений, качества оказываемой медицинской помощи и прохождения ежегодных обследований. При соблюдении этих условий пациенты могут жить полноценной и качественной жизнью. В таких случаях ее продолжительность близка к продолжительности жизни людей без особенностей здоровья.
Что включает в себя облегчение симптомов БЭ
Симптоматическое лечение имеет важнейшее значение для людей с БЭ. Обычно оно включает ежедневный уход за непораженной кожей, за кожей с пузырями и ранами, а также обезболивание.
Пациентам с более тяжелыми типами БЭ требуются дополнительные процедуры, в том числе прием препаратов железа, переливание эритроцитарной массы и альбумина — белка, находящегося у данных пациентов в большом дефиците. Именно от его количества в организме зависит скорость заживления кожи, в том числе и после хирургических вмешательств. Пузыри поражают слизистую и заживают только через образование рубца, который сужает просвет органа или ограничивает его функцию. Хирургические операции позволяют освободить сросшиеся пальцы и открыть пищевод для нормального поступления пищи.
Как выглядит симптоматическое лечение при БЭ
Характер и стоимость ухода за ранами зависят от многих факторов, в том числе имеют значение:
- форма БЭ;
- возраст пациента;
- общее восприятие;
- текущее состоянии кожи;
- состояние питания;
- домашнее окружение;
- наличие качественных перевязочных материалов.
Основное лечение подразумевает использование атравматического материала, с которым при снятии не будет отходить кожа.
Основные этапы перевязки:
Тщательная подготовка к перевязке со всеми необходимыми материалами и доступным механизмом утилизации старого перевязочного материала сокращает длительность перевязки и снижает риск возникновения инфекции.
Все лица, участвующие в смене повязок, должны в обязательном порядке проводить предварительную дезинфекцию рук.
Оценка состояния кожи
Все пузыри на коже необходимо вскрывать, затем очищать образовавшиеся раны, при инфицировании — лечить.
При перевязке на кожу сначала накладывают сетчатую или губчатую повязку, затем для впитывая экссудата и излишков крема — промежуточную, после этого — фиксирующую повязку.

Что важно при выборе перевязочных средств и уходе за кожей при БЭ
Интенсивность ухода за ранами определяется в первую очередь формой БЭ. При простой форме, когда пузыри и раны возникают на отдельных участках тела, а не повсеместно, перевязка проходит гораздо быстрее, чем при более тяжелой дистрофической форме, требующей обработки повязками большей части кожи.
К счастью, в настоящее время есть множество перевязочных средств и медикаментов, которые подходят для ухода за ранами при буллезном эпидермолизе. Однако найти «правильный» материал все еще нелегко. То, что подходит для одного больного, может оказаться совсем неподходящим для другого. Поэтому даже при одной и той же форме БЭ перевязочный материал и уходовые средства подбираются индивидуально. Оценка такого соответствия должна всегда производиться совместно с врачом.
При выборе перевязочных средств необходимо также учитывать их доступность на местном уровне, ведь в разных странах для этих целей разработаны и доступны разные продукты. Кроме того, на перевязочные средства требуется значительная сумма. Так, в России на уход за кожей человека-бабочки ежемесячно нужно от 50 до 400 тыс. руб. Сегодня наше государство не всегда покрывает эти траты, а при обеспечении за счет бюджета отдает предпочтение менее дорогостоящим материалам.
Еще одним важным аспектом в лечении больного БЭ является гигиена и уход за участками кожи с рубцами, без пузырей и ран. Такой коже в первую очередь требуются восстановление и увлажнение. Чем лучше и качественнее увлажнена кожа, тем меньше выражены рубцы, меньше зуда и ниже риск инфицирования. Увлажняющие крема необходимо наносить на открытые участки кожи два-три раза в день.
Может ли легкая форма буллезного эпидермолиза перейти в тяжелую
Нет, поскольку причины, вызывающие одну форму БЭ, отличаются от причин возникновения других типов этого заболевания. Они, по сути, являются отдельными состояниями, поэтому одна форма не может перерасти в другую.
Буллезный эпидермолиз

Буллезный эпидермолиз (сокр. БЭ) – это понятие объединяющее целую группу редких наследственных болезней, имеющих генетическую и клиническую гетерогенность. Чаще всего патологические процессы происходят в кератиноцитах и приводят к образованию пузырей с доброкачественным течением, возникновению эрозивных участков кожи и слизистых оболочек, повышенной ранимости и чувствительности кожных покровов к любым незначительным механическим травмам, развитию «механобуллезной болезни», которая еще называется синдром Бабочки. Встречаемость разных видов пузырчатки – всего 1 случай на 30-100 тыс. человек.
Ввел понятие буллезного эпидермолиза немецкий дерматолог Генрих Кёбнер еще в 1886 г., хотя подобные случаи кожных заболеваний встречались и ранее.
Продолжительность жизни при буллезном эпидермолизе
Несмотря на стремительное развитие всемирной медицины продолжительность жизни у пациентов с врожденным эпидермолизом – «детей-бабочек» чаще всего не высокая. Все зависит от подтипа заболевания, глубины и обширности патологических поражений. Простой буллезный эмидермолиз при правильном уходе и комплексной терапии протекает легче, с возрастом минимизируется количество пузырей и прогноз для таких пациентов благоприятный. Другие типы генодерматозной болезни, особенно при присоединении инфекций, чаще всего стафилококковой или стрептококковой, при развитии системных осложнений и сепсиса чаще всего приводят к летальному исходу в ранние годы жизни детей.
Патогенез
Патология выявляется с рождения – на коже имеются пузырчатые образования с серологическим содержимым, которые при вскрытии примерно через 2-3 дня оставляют долго незаживающие эрозии, чаще всего приводящие к развитию атрофичной рубцовой ткани, гипер- либо гипокератоза, повторному развитию пузырей. Известны случаи манифестации болезни и в более позднем возрасте – в младенчестве, раннем детстве и даже юношестве.
Поражение кожи — возникновение язв и пузырей вызывают любые механические воздействия, поэтому больше всего страдают участки с наиболее частым трением – подмышки и различные складки, места прилегания одежды и лямок.
В основе патологии лежат мутации генов, кодирующих белки различных слоев кожи – базальных мембран, дермы. При этом происходит нарушение баланса в системе ферментов и ингибиторов, белки становятся объектом атаки защитных систем организма – ферментативного цитолиза, вызывающего отек цитоплазмы, разрыв клеточных оболочек и как следствие — нарушений межклеточных связей, структуры эпидермиса с образованием внутри- либо субэпидермальных булл и везикул.
Классификация
Изучая на ультраструктурном уровне морфологию кожи (количество, виды таких структур кожи как нити кератина, десмосомы, гемидесмосомы, якорные нити, якорные волокна и пр.) и локализацию пузырчатых образований в слоях эпидермиса. Выделяют три основных типа механобуллезного эпидермолиза, которые подразделяются на более чем 30 подтипов в зависимости от фенотипических и генетических особенностей больных, а также типа наследования:
- Простой тип (эпидермолитический согласно классификации Пирсона 1962 г.) – наиболее распространненный, встречается в 75% случаев и характеризуется образованием пузырей в верхних слоях эпидермиса на коже стоп, кистей рук, в тяжелых случаях – всего тела. Разделен на 2 основных подтипа супрабазальный, при котором белками-мишенями становится плакофилин-1, десмоплактин и пр., а также базальный подтип, вызывающий изменения структуры альфа-6/бета-4 –интегрина, причем кожные нарушения могут быть локализованными, генерализованными (пузыри возникают группами) и в виде пятнистой пигментации. Наиболее часто встречающимися клиническими формами является летальный акантолитический, поверхностный, с мышечной дистрофией, с атрезией привратника, огнасский, мигрирующий кольцевидный и т.д.
- Пограничный тип (люцидолитический) ведет к формированию пузырей в области светлой пластинки базальной мембраны и встречается в форме подтипа Херлитца, нарушающего строение ламининов332 и 5, а также других подтипов вовлекающих в патогенез не только ламинины, но и коллаген XVII типа и α альфа-6/бета-4 –интегрин. Кроме того, выделяют клиническую форму инверсионную, с поздним началом, в виде ларинго-онихо-кутанного синдрома.
- Дистрофический тип (дермолитический) – поражает верхнюю часть сосочкового слоя толщи дермы, который бывает доминантного и рецессивного подтипа и развивается за счет нарушения структуры коллагена четвертого типа. Он бывает генерализованным, периферическим, претибиальным, центрипетальным, пруригинозным, поражающий только ногти, инверсным.
- Отдельной формой, наиболее редкой и малоизученной считается синдром Киндлера, так как пузыри могут образовываться в разных слоях эпидермиса и вызвано это нарушением структуры белков – киндлинов-1. Они выявляются сразу при рождении ребенка, чаще всего на кожных покровах рук и ног. В последствие может наблюдаться возникновение дистрофических изменений ногтей, развитие кариеса, пародонтита, различных заболеваний ротовой полости, ЖКТ, оболочек глаз, мочеполовой системы. С возрастом количество новообразующихся пузырей сокращается, но кожа все равно остается тонкой, легко ранимой, чувствительной, капиллярная сетка располагается слишком близко к поверхности эпидермиса.
Причины буллезного эпидермолиза
Основные причины возникновения патологии — мутации более чем в 10 генах, отвечающих за кодирование белков различных слоев кожи, чаще всего в 75% — это гены KRT5 и 14, LAMC2, LAMA3, LAMB3, COL17A1. Как указывает онлайн-ресурс Википедия почти для каждого из установленных подтипов БЭ удалось выявить мутации в определённых генах, среди них чаще всего встречаются:
- миссенс точечные мутации;
- нонсенс точечные мутации;
- делеции и инсерции – потери и соответственно вставки участков хромосом;
- мутации сдвига рамки считывания;
- сплайсинг.
Тип наследования буллезного эпидермолиза встречается как аутосомно-рецессивный (наиболее часто), аутосомно-доминантный, так и однородительская дисомия, и соматический мозаицизм.
Факторы, провоцирующие врожденный буллезный эпидермолиз
Врожденный синдром Бабочки может развиться даже у здоровых родителей, не несущих мутированные гены. Спонтанные мутации возникают внутриутробно и причиной тому становится:
- вредные привычки беременной женщины – курение, злоупотребление алкоголем;
- хаотичный прием лекарственных препаратов и прочие тератогенные факторы.
Симптомы
Симптоматика буллезного эпидермолиза связана с нарушением структуры кератиноцитов и проявляется в виде:
- жжения, боли в местах поражений и облегчения при опорожнении пузырей;
- зуда во время заживления вскрывшихся пузырей – отслоения подсохшей крышки везикулы, что в дальнейшем может вызвать шелушение и пигменатцию;
- развития участков кожи с эрозией;
- повышения чувствительности и ранимости кожных покровов к любым механическим повреждениям;
- спонтанного возникновения напряженных пузырей, содержащих прозрачную бесцветную жидкость или геморрагическое содержимое.
Патология обычно обнаруживается сразу при рождении, ведь тонкая и «ломкая» кожа легко травмируется даже от прохождения по родовым путям. При этом кожные поражения имеют разную степень тяжести и распространенности даже в пределах семьи. Множественные распространенные пузыри могут вызвать смерть новорожденного, особенно при присоединении вторичной инфекции. Обострения обычно припадают на летнее – теплое время года, с возрастом распространенность пузырей минимизируется.
Дополнительные симптомы простого БЭ
Простые эпидермолитические поражения обычно возникают на кистях рук и на стопах либо могут покрывать всё тело. Кроме того может наблюдаться:
- развитие ладонно-подошвенного гиперкератоза распространённого или сливного вида;
- дистрофические изменения ногтевых пластинок; ;
- милии;
- гипер- либо гипопигментация;
- задержка роста.
Заживление пузырей чаще всего происходит без рубцевания, иногда даже напоминает простой герпес, но при этом очень высока вероятность рецидивов.
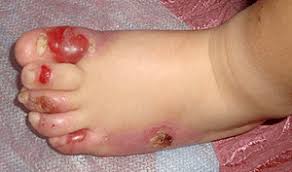
Простой буллезный эпидермолиз
Особенности течения пограничного БЭ
Этот тип отличается развитием патогномоничного симптома – возникновения участков обильной грануляционной ткани на различных частях тела (как на фото буллезного эпидермолиза пограничного типа), например, локализованной симметрично вокруг рта, в средней части лица, вокруг носа, на верхней части спины, в подмышечных впадинах или ногтевых валиках. Патология может привести к ониходистрофии и даже полной утрате ногтевых пластинок, большому количеству милий, серьёзных рубцов на теле, в том числе рубцовой алопеции, корок на коже, язв в ротовой полости, гипоплазии эмали и тяжелому кариесу зубов. Среди возможных системных осложнений обычно выявляют полиэтиологическую анемию, задержку роста, эрозии, кожные везикулярные высыпания и отслоение, стриктуры в желудочно-кишечном тракте. Риск их развития повышается в зависимости от площади, степени деструктивности и глубины кожных поражений.
Пограничнй буллезный эпидермолиз
Пограничный буллезный эпидермолиз характеризуется крайне высокой смертностью преимущественно в первые годы жизни, вызванной прекращением прибавки в весе, сепсисом, пневмонией или обструкцией трахеи и гортани.
Внекожные проявления дистрофического БЭ
Помимо генерализованных кожных поражений — рецидивирующего образования пузырей, эрозий, милий, атрофического рубцевания и утраты ногтей «синдром Бабочки» вызывает:
- нарушения работы желудочно-кишечного тракта;
- поражение мочеполового тракта и внешних глазных оболочек;
- хроническую анемию; ;
- задержку роста;
- уплотнение, дистрофию и даже утрату ногтевых пластинок;
- контрактуры различных суставов конечностей – локтевых, коленных, лучезапястных, плюсне-предплюсневых, плюснефаланговых и т.д.;
- повышенный риск развития новообразований, к примеру, агрессивной плоскоклеточной карциномы.
Дистрофический буллезный эпидермолиз
Анализы и диагностика
Наибольшее значение в постановке диагноза играет биопсия — исследование образцов кожи при помощи трансмиссионных электронных микроскопов, позволяющих осуществить визуализацию и провести полуколичественный анализ различных структур эпидермиса. Благодаря доступности моноклональных и поликлональных антител против белков разных слоев эпидермиса, участвующих в патогенезе буллезного эпидермолиза на сегодняшний день всю большую популярность завоёвывают иммуногистологические методы.
Иммуногистохимический метод исследования и метод непрямой иммунофлюорисценции позволяют определить состояние экспрессии структурных белков с наследственными дефектами в клетках кожи — кератиноцитах и базальных мембранах (основы эпителия, выполняющей барьерную и трофическую функцию), а также схему распределения протеидов в ранее образованных или искусственно спровоцированных пузырях, в том числе — их глубину локализации.
Благодаря современным методам ДНК-диагностики удается быстро классифицировать разновидность патологии, выявить структурные белки, подвергшиеся мутации и составить клинический прогноз. Инновационный метод генетического анализа – прямое секвенирование дает возможность выявить мутации, их тип и локализацию, достоверно подтвердить диагноз.
Помимо этого важную роль играет сбор семейного анамнеза и истории болезни пациента, комплексное обследование всего организма, проведение лабораторных анализов.
Лечение буллезного эпидермолиза
Наиболее эффективным и распространённым на сегодняшний день считается превентивное и симптоматическое лечение, так как радикальной терапии для излечения любого из подтипов БЭ до сих пор разработано не было, хотя точно поставленный диагноз дает возможность существенно повысить качество жизни и снизить вероятность рецидивов.
Наиболее перспективными считаются направления лечения, способные препятствовать образованию или заменять белки кожи неправильной структуры:
Люди-бабочки: что такое буллезный эпидермолиз и как наука ищет способы его лечить
Как устроена наша кожа и из-за чего возникает болезнь, при которой кожа становится очень хрупкой и может повредиться от любого прикосновения? Как живут люди, которых называют «бабочками», и чем медицина может им помочь?
Вместе с информационно-просветительским гуманитарным проектом «12 месяцев» мы продолжаем серию материалов о редких (орфанных) генетических заболеваниях и жизни людей с ними.
Читайте в январе рассказ о буллезном эпидермолизе, который встречается у одного из 100 тысяч человек, а также историю юриста Игоря Чувствинова.
Что важно знать о буллезном эпидермолизе
Буллезный эпидермолиз (БЭ) — это группа генетических заболеваний, при которых даже незначительное механическое воздействие на кожу и слизистые оболочки приводит к образованию пузырей. Когда эти пузыри разрываются, на их месте остаются болезненные раны, которые быстро инфицируются и с трудом поддаются лечению.
Болезнь сильно снижает качество жизни людей и требует сложного и дорогостоящего ухода. Нередко проявления заболевания выражены настолько, что пациенты полностью зависят от своих близких.

Источник: сайт фонда «Дети-бабочки»
Точно посчитать частоту встречаемости буллезного эпидермолиза сложно. По данным Национального института здоровья США, заболеваемость БЭ составляет примерно 1 случай на 100 тысяч человек в общей популяции и почти 2 случая на 100 тысяч новорожденных. В России, по оценкам фонда «Дети-бабочки», который помогает детям с буллезным эпидермолизом, с этим заболеванием проживает около 2000-2500 человек.
Почему возникает и как проявляется буллезный эпидермолиз?
Кожа человека состоит из нескольких слоев клеток — кератиноцитов. Они соединяются и удерживаются вместе с помощью специальных белков, основные из которых — это кератины, коллаген, ламинин и киндлин. Мутации в генах, необходимых для их синтеза, приводят к нарушению функции этих белков. Это приводит к тому, что даже при небольшом натяжении или давлении на кожу ее слои отходят друг от друга и в этом месте образуются пузыри.
Основные проявления БЭ связаны с кожей, однако болезнь затрагивает весь организм. Так, наиболее часто поражаются слизистые оболочки желудочно-кишечного тракта. В ротовой полости со временем образуются эрозии и пузыри, а в пищеводе — сужения, значительно затрудняющие глотание. Кроме того, нередки случаи нарушения проходимости кишечника, анальных трещин и запоров.
Болезнь поражает и глаза: к ее последствиям относится хроническое воспаление, язвы на роговицах. Способность к самообслуживанию чрезвычайно сильно ограничивает ситуация, когда срастается кожа пальцев.
Еще одна опасность для людей с БЭ — повышенный риск рака кожи, который увеличивается с возрастом.
Можно ли вылечить буллезный эпидермолиз?
Основные способы лечения буллезного эпидермолиза — это ведение образа жизни, уменьшающего вероятность травмирования кожи, и использование специальных дорогостоящих повязок. Однако уже давно у врачей и исследователей появилось желание воздействовать на причину заболевания — неспособность клеток плотно соединяться друг с другом.
Начиная с конца 1980-х годов предпринимались разные попытки лечения, основанные на методах клеточной терапии (то есть терапии стволовыми клетками). Их цель — улучшить течение болезни в тех ситуациях, когда обычное лечение не помогало. Однако в большинстве случаев эффект был выражен слабо и длился очень недолго.
Первым подходом клеточной терапии стала попытка трансплантации пациентам с БЭ кератиноцитов, которые были взяты из непораженных участков кожи. Это дало ограниченный и непродолжительный эффект, который, по-видимому, был обусловлен временным уменьшением воспаления в зоне трансплантации.
Другой подход основывался на внутривенном введении стволовых клеток костного мозга и мезенхимальных стромальных клеток (МСК), способных мигрировать в зоны повреждения. Исследователи предполагали, что эти клетки, трансплантированные от здоровых доноров, синтезируют белки, необходимые для образования нормальной кожи. Однако, как и в предыдущем случае, эффект этого лечения был успешным лишь отчасти.
При использовании стволовых клеток костного мозга наблюдалось временное улучшение течения болезни. Однако несколько пациентов в процессе клинического исследования умерли от осложнений, связанных с подготовкой к трансплантации.
В случае использования мезенхимальных стромальных клеток (МСК) столь драматичных побочных эффектов не было. Но эффект от лечения — ускорение заживления ран и улучшение общего самочувствия — по-прежнему длился не более полугода.
Из-за сложностей при получении стволовых клеток костного мозга и МСК, ученые обсуждают возможность использования индуцированных плюрипотентных стволовых клеток (ИПСК). ИПСК — это любые клетки, которые в лабораторных условиях приводят в т.н. плюрипотентное состояние (когда клетка становится недифференцированной, какой была в эмбриональном периоде развития). Из него они могут стать почти любой другой клеткой, в том числе стволовой клеткой костного мозга, МСК или сразу фибробластом или кератиноцитом. Однако процесс получения клеток из ИПСК пока технически несовершенен, поэтому исследования таких клеток проводятся пока только в условиях лабораторий.
Есть ли надежда на генно-клеточную терапию?
Может сложиться впечатление, что введение стволовых клеток костного мозга и мезенхимальных стромальных клеток (МСК) в системный кровоток малоперспективно из-за технического несовершенства этого подхода и кратковременности терапевтического эффекта. Тем не менее дальнейшие исследования в этом направлении необходимы, потому что на сегодняшний день это единственный подход, потенциально позволяющий действовать на внекожные проявления болезни.

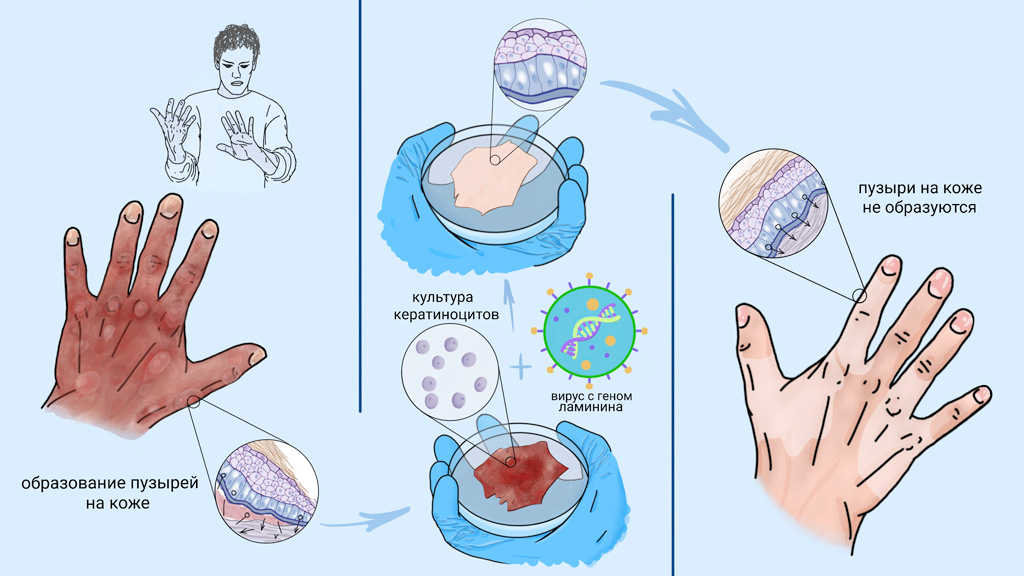
О первом успехе генно-клеточной терапии буллезного эпидермолиза стали говорить в 2006 году, когда группа итальянских исследователей смогла восстановить кожу пациента с пограничным буллезным эпидермолизом. Эта форма заболевания появляется при мутации в гене белка ламинина. Для лечения пациента исследователи выделили из неповрежденных участков его кожи кератиноциты, в которые ввели здоровый ген ламинина. Далее в лабораторных условиях из этих кератиноцитов вырастили тонкие пласты ткани, которые трансплантировали в поврежденные участки кожи. Через полгода кожа в зоне трансплантации не отличалась от нормальной и не требовала повторных вмешательств.
В 2015 году успех этого подхода закрепил опыт по трансплантации практически всего эпидермиса семилетнему мальчику, который к моменту первой трансплантации утратил около 80% кожных покровов.
В результате лечения у пациента сформировалась полностью здоровая и функциональная кожа. Сейчас трансплантированная кожа пациента почти не отличается от здоровой. Он ведет обычную жизнь и даже занимается спортом, хотя до лечения из-за боли практически не мог двигаться, нормально питаться и нуждался в постоянном обезболивании.
-
Сегодня эта технология используется в трех препаратах, проходящих клинические исследования — EB-101, LZRSE-Col7A1 и FCX-007. Другой препарат, проходящий клинические исследования — B-VEC. Его преимущество — возможность непосредственного введения в кожу пациентов, что позволяет миновать затратный по времени этап выделения и выращивания клеток пациента.
«Заболевание переориентировало меня на книги»: как жить с буллезным эпидермолизом
26-летний Игорь Чувствинов рассказал, как ему удается учиться, работать, дружить и чувствовать себя счастливым состоявшимся человеком, несмотря на ограничения из-за болезни.

О детстве и постановке диагноза
Дети с буллёзным эпидермолизом нередко имеют слабую кожу уже на момент рождения и получают травмы при родах. Мой случай не был исключением. Но до 3-х лет родителям не понимали, что со мной происходит.
Были две сложности в постановке диагноза: во-первых, врачи первоначально хотели поставить пузырчатку (аутоиммунное заболевание, которое характеризуется появлением пузырей на коже и слизистых оболочках), потому что симптомы этих заболеваний похожи. Во-вторых, врачи принимали проявления болезни за временные явления и ждали, что они пройдут сами, но они не проходили.
Я родился в 1995-м году, и понятно, что всем тогда было не до редких заболеваний. О них не говорили в СМИ, не было профильных фондов. И врачи оказывались в очень сложном положении, видя меня.
На вопрос: «Чем лечиться?» дерматологи отвечали: «Вы столько прожили с этим заболеванием, вы знаете о нем больше, чем я».
О влиянии болезни на жизнь
Я не знаю жизни без этого заболевания, поэтому мне сложно сравнить «до» и «после». Но если смотреть на мою жизнь и жизнь других людей, то, конечно, отличия есть.
Во-первых, я намного чаще наблюдался у врачей.
Во-вторых, точно могу сказать, что мое заболевание повлияло на спектр моих хобби. Я никогда не занимался опасным спортом, не катался на велосипеде, не играл в футбол, не ходил далеко гулять. В детстве это могло быть очень травматичным для моих ног.
Я больше домосед. Заболевание переориентировало меня на книги, фильмы, компьютерные игры и прочее, что не связано с активностью, способной привести к травмам. При этом я довольно общительный человек, поэтому не могу сказать, что болезнь мешала или мешает мне в социализации.
В-третьих, мой диагноз влияет на то, что я ем: я не употребляю продукты, которые могут вызвать аллергическую реакцию. После чего кожа может начать чесаться. И есть риск нарваться на круговорот болячек в природе, когда они уже не заживают, потому что постоянно их расчесываешь. Естественно, что в детстве за этим следили родители, а потом я стал сам видеть связь между употреблением какого-то продукта и последствиями, которые оно вызывает.
И если раньше меня печалило, что мне 14 лет, а я ни разу не пробовал колу, то потом я стал думать: «Ну и хорошо, что не попробовал. А то, кто знает, что было бы?». Я питаюсь, как и остальные члены моей семьи, просто исключив вредные для меня продукты.
Абсолютно все, что связано с применением физической силы, вызывает у меня проблемы. Например, мыть посуду я могу только в перчатках, но на деформированную кисть перчатка не налезает. Если делать это без перчаток, может быть какая-то нехорошая реакция на моющее средство, а кожа на руках будет сохнуть еще сильнее. Я не могу самостоятельно открыть бутылку воды, потому что из-за этого механического воздействия мои ладони травмируются. Я могу открыть не любую дверь. Если дверная ручка шарообразной формы, да ещё и с рельефом, травмы не избежать. Я не могу преодолевать большие расстояния пешком. Чем дольше я иду, тем сильнее это травмирует мои ноги. Даже небольшой выступ на асфальте может быть достаточным для появления раны на стопе. И так со многими действиями.
Но я не совсем беспомощный — с технологиями проблем нет. Я пишу, пользуюсь клавиатурой и мышкой. В работе мне приходится часто и быстро печатать большое количество текста, в том числе на телефоне. С этим я справляюсь.
Об отношении к болезни
Мое заболевание никогда не было для меня чем-то, что заставляло бы меня отчаиваться и падать духом. Скорее, я воспринимал его как само собой разумеющийся факт. Например, как тот, что солнце встает на востоке и заходит на западе.
Я осознаю свои ограничения. Тут вопрос не в том, осознаю ли я их, а в том, как я к ним отношусь. С детства в силу возраста и характера я переживал из-за этого не так сильно, как мог бы. А сейчас мне 26 лет, и я привык к этим ограничениям.
Многие мои сверстники водят автомобиль, а я нет. Когда мне было 18, меня это сильнее трогало, сейчас мне это глубоко индифферентно. Не вожу — и не вожу. Может, и к лучшему. Ни в кого не врежусь и не буду за это привлечен к ответственности. Хорошо, что не ем всякую химозу, зато у меня здоровый желудок. Какое-то такое отношение. С годами стало проще.
Понятно, что некоторые проявления болезни доставляют дискомфорт, но я отношусь к этому со смирением и осознанием того, что если что-то нельзя, то и ладно, живем дальше. С этим можно жить.
И главный вопрос «Как жить?». Отчаявшись? Не ставя перед собой никаких целей? Жить, как получится? Или, даже несмотря на заболевание, стараться чего-то достичь, кому-то помочь? Мне повезло, что сейчас не Средние века, и я вижу много возможностей для самореализации и социализации даже при наличии таких существенных отклонений, как у меня.
О друзьях, учебе и жизни в обществе
Никакого буллинга в мою сторону никогда не было, по крайней мере активного. Я учился в обычной средней общеобразовательной школе, потом поступил в университет. И везде у меня появлялись друзья — общения в моей жизни немало.
Я юрист, сейчас одновременно учусь в аспирантуре и прохожу стажировку в крупной компании. Все выходные и праздники заполняю дополнительной учебой.
Два моих главных хобби с детства — чтение книг и компьютерные игры.
Люблю читать психологов, один из моих любимых — Виктор Франкл. Его «Скажи жизни да» — это, можно сказать, моя настольная книга.
Из последних впечатливших меня книг, — «Игры, в которые играют люди» Эрика Берна. Однако чем старше я становлюсь, тем меньше художественной и больше профессиональной литературы я читаю.
Как я уже говорил, в обычной жизни я испытываю ограничения там, где у других людей их нет. Однако не могу сказать, что испытываю какую-то радость, когда у меня получается что-то, что получается у других. Обычно наоборот: я испытываю радость, когда я делаю что-то, что не могут другие. Вот тогда да! Во многом это касается профессиональных успехов.
О техническом прогрессе, который помогает людям-бабочкам
Кто знает: возможно, лет через 5-10 я и машину смогу водить. Например, еще 5 лет назад у меня не было никакой возможности поставить коронки на зубы. До 8 лет мои зубы в принципе лечить не хотели. «Мы не можем это лечить. Ваш рот плохо открывается. Мы боимся вас травмировать».
Был случай, когда в одной клинике врач выбежала из кабинета с криками: «Я не хочу в тюрьму!». Потому что мое заболевание не только про травмы кожи, но и про травмы слизистых оболочек.
Недавно фонд «Дети-бабочки» обучил врачей в клинике челюстно-лицевой хирургии имени Сеченова. И сейчас мне поставили на некоторые зубы коронки, появились технологии, которые это позволяют. Это та реальность, которая придает мне оптимизм. Технический прогресс на месте не стоит, и он работает на меня.
Однако я бы не согласился участвовать в клиническом исследовании нового препарата от своей болезни. Несмотря на все ограничения, которые у меня есть, я выбираю свою нынешнюю жизнь со всеми неудобствами, а не рискованные методы лечения.
Отдельно я хочу выразить благодарность фонду «Дети-бабочки». Он вносит существенный вклад в мою жизнь и в жизнь других людей с буллезным эпидермолизом — до него нам было намного сложнее.
Благодаря просветительской работе фонда, намного больше врачей знают, что есть такая болезнь. И значит, люди с таким диагнозом могут к ним прийти, быть понятыми и получить помощь.
Синдром бабочки

Заболевание, связанное с генетической аномалией, из-за которой кожа человека не выполняет своей защитной функции, называют синдромом бабочки. Выбор наименования объясняет состояние кожи – она становится ранимой, словно крылья бабочки, которая не полетит после прикосновения к ней человеческих пальцев.
Буллезный эпидермолиз – это
Буллезным эпидермолизом называют заболевание кожи, при котором механические воздействия, даже легкие прикосновения к телу становятся причиной образования волдырей, пузырей и эрозии на коже.
Исследования показали, что это заболевание передается по наследству, но доминантность или рецессивность этой болезни не установлена из-за множества разновидностей синдрома бабочки, одни из которых передаются потомству при разных условиях, в другие передаются через поколение или без точной периодичности.
Чрезмерная нежность кожного покрова приводит к тому, что человек не может вести нормальный образ жизни: любое касание к швам на одежде, предметам мебели, тактильные контакты с другими людьми приводят к разрыву эпидермиса или образованию пузырей. Капилляры, расположенные близко к поверхности кожи, лопаются. Из-за чего возникают необильные кровотечения, которые трудно остановить из-за большой площади поражения.
Кроме того, обработка таких ран тоже представляет определенную сложность, потому что любые контакты только усугубляют состояние раны. В особенно тяжелых случаях можно спастись только если купить спрей с антисептиком. Промыть раны или протереть их ватным диском с обеззараживающим раствором нельзя.
Причины возникновения синдрома бабочки
Происхождения разных типов заболевания до сих пор до конца не изучены. Это существенно усложняет проработку тактики лечения и предупреждения болезни. Простой и самый распространенный тип буллезного эпидермолиза возникает при мутации всего двух генов. Они имеют имена KRT5 и KRT14. Но в результате систематизации информации, полученной в после исследования каждого случая, было выявлено, что только в 75% случаях виновата была мутация генов. При этом в кожных слоях нарушается работа системы отношения ферментов и ингибиторов. В результате, под удар попадают некоторые белки. При основном типе буллезного эпидермолиза повреждению подвергаются протеины базальной мембраны и десмоплакин.
При прикосновении к коже происходит выброс ферментов, которые разрушают эти белки. Это и становится причиной начала образования пузырьков разного размера. При легкой степени заболевания эти пузыри не разрушают кожу сильно глубоко, но при тяжелых течениях возникают волдыри, разрушающие полностью все слои эпидермиса.
Другой тип синдрома бабочки связывают с мутацией двух других генов: LAMB3 и LAMA3. В редких случаях вместе с ними изменениям подвергаются и другие. Наследование этого типа описывается как рецессивное. От атак со стороны ферментов в этом случае страдают другие белки – разновидность коллагена и ламинина. Их прямое назначение заключается в поддержке структуры самых нижних слоев кожи, поэтому их повреждение отличается специфической клинической картиной – такое течение болезни наиболее сложно переносимо из-за очень глубокого поражения эпидермиса.
Рассматривая дистрофический тип синдрома бабочки, следует отметить, что это менее тяжелое заболевание. Оно обусловлено поражением такого типа коллагена, которые содействует крепости соединительной ткани в слоях кожи. Его разрушение ведет к некоторым высыпаниям и эрозийными повреждениями. Но именно это заболевание считают одним из самых опасных, потому что разрушения этого типа коллагена отражается на поражении других органов, в частности – слизистой оболочки, выстилающей органы дыхания и весь желудочно-кишечный тракт. Рубцы, образовывающие после заживления волокон, часто перерождаются в онкологические заболевания.
Подводя итог, можно отметить, что причина хрупкости кожи, независимо от типа буллезного эпидермолиза, связана напрямую с разрушением белков из-за нарушений ферментарной активности непосредственно в тканях. Болезнь разбили на типы, чтобы объяснять и систематизировать локализацию образования пузырей.
Симптомы буллезного эпидермолиза
Главным характерным симптомом заболевания является появление пузырей на поверхности кожи при малейшем ее касании. Однако следует отметить, что разные типы синдрома бабочки по-разному отражаются на коже.
Существует разновидность буллезного эпидермолиза, при котором волдыри появляются только на определенных участках конечностей. Также существует такой тип болезни, который вызывает лишь мелкую сыпь, но из-за площади ее поражения тяжесть течения не становится легче.
Как правило, генетические нарушения выявляются с младенческого возраста. Что очень усугубляет состояние ребенка. Он, не понимая сложности ситуации, и не контролируя еще свои движения, может сам себе наносить увечья. Основное число заболевших тяжелыми формами синдрома бабочки не доживают и до 5 лет.
Выжившие становятся инвалидами. Они могут приспособиться к жизни, сильно при этом ограничивая себя – исключая тактильные контакты, посещения общественных мест, а часто даже выход из дома. У таких больных, как правило, прогрессирующий кариес, который к определенному возрасту вовсе лишает зубов из-за истончения эмали зубов. Ногти у них не просто становятся ломкими – они выпадают. Кожа и кутикула не способны их удерживать.
Осложнения буллезного эпидермолиза
В связи с тем, что заболевание напрямую связано с поражением кожных покровов широкой площади, первым и самым главным осложнением является присоединение инфекции. В отрытые раны легко занести болезнетворную флору, которая может стать причиной нагноений.
При обширных поражениях глубоких слоев возникает болевой шок. Лечение буллезного эпидермолиза в основном направлено на симптоматику и снижение риска возникновения сепсиса. Раны обрабатываются щадящими анисептическими растворами, а внутрь, в зависимости от возраста больного, могут приниматься препараты группы нестероидных (или стероидных) противовоспалительных средств, обладающих обезболивающим действием.
Все представленные на сайте материалы предназначены исключительно для образовательных целей и не предназначены для медицинских консультаций, диагностики или лечения. Администрация сайта, редакторы и авторы статей не несут ответственности за любые последствия и убытки, которые могут возникнуть при использовании материалов сайта.

